cílem tohoto tutoriálu je seznámit vás se zpracováním sekvenčních dat nové generace v galaxii. Tento tutoriál používá covid-19 variantu volání z dat Illumina, ale nejde o variantu volání samo o sobě.
po dokončení tohoto kurzu budete vědět:
- Jak najít data v SRA a přenos této informace do Galaxie
- Jak provést základní NGS zpracování dat v Galaxy, včetně:
- Kontrola Kvality (QC) z Illumina dat
- Mapování
- Odstranění duplikáty
- Varianta volání s
lofreq - Varianta anotace
- Pomocí datových souborů sbírek
- Import dat do Jupyter
### Agenda>> V tomto tutoriálu, budeme pokrývat:>> 1. Toc> {: toc}> {: .agenda}# # dvě cesty přes tento tutoriálvytvořili jsme dvě trajektorie, které můžete sledovat v tomto tutoriálu.1. ** Trajektorie 1 * * – Začněte s SRA NCBI a vyhledejte dostupné přístupy → Start (#the-sequence-read-archive)2. ** Trajektorie 2 * * – bypass NCBI je SRA a začít s Galaxy přímo. → Start (#back-in-galaxy)doporučujeme začít s * * trajektorie 2**.# Sekvence Číst ArchiveThe (https://www.ncbi.nlm.nih.gov/sra) je primární archiv *nesmontované čte* (https://www.ncbi.nlm.nih.gov/). SRA je skvělým místem pro získání sekvenčních dat, která jsou základem publikací a studií.Tento tutoriál popisuje, jak získat sekvenční data ze SRA do Galaxy pomocí přímého spojení mezi nimi.> # # # comment Comment>> uslyšíte také SRA označovaný jako * Short Read Archive*, jeho původní název.> {:.komentář} # # Přístup k SRASRA lze dosáhnout buď přímo prostřednictvím svých webových stránek, nebo prostřednictvím panelu nástrojů na Galaxy.> ### komentář Komentář>> Zpočátku panelu nástrojů možnost pro přístup SRA existuje pouze na (https://usegalaxy.org/). Podpora pro přímé připojení k SRA bude zahrnuta v 20.05 vydání Galaxy{: .komentář}> ### hands_on Hands-on: Prozkoumat SRA Entrez>> 1. Jdi do svého Galaxy instance volba, jako jeden z (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au), nebo jakékoli jiné. (Tento návod používá usegalaxy.org).> 1. Pokud vaše historie ještě není prázdná, než začít novou historii (viz (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) pro více informací o historii galaxií)> 1. ** Klikněte* * „získat Data“ v horní části panelu nástrojů.> 1. ** Klikněte * * ‚Sra Server‘ v seznamu nástrojů uvedených v části „Získat Data“`> tím získáte (https://www.ncbi.nlm.nih.gov/sra) – můžete také začít přímo z SRA. V horní části stránky se zobrazí vyhledávací pole. Zkuste hledat něco, co vás zajímá `například „dolphin“ nebo „ledviny“ nebo „dolphin ledviny“ a pak* * klepněte na tlačítko * * na tlačítko „Hledat“.>> tím se vrátí seznam * Sra experimentů*, které odpovídají vašemu vyhledávacímu řetězci. Experimenty SRA, známé také jako * SRX entries*, obsahují sekvenční data z konkrétního experimentu, stejně jako vysvětlení samotného experimentu a dalších souvisejících dat. Vrácené experimenty můžete prozkoumat kliknutím na jejich jméno. Viz (https://www.ncbi.nlm.nih.gov/books/NBK56913/) v (https://www.ncbi.nlm.nih.gov/books/n/helpsrakb/) pro více informací.>> Když zadáte text ve SRA search box, který používáte (https://www.ncbi.nlm.nih.gov/sra/docs/srasearch/). Entrez podporuje jak jednoduché textové vyhledávání, a velmi přesné vyhledávání, které zkontrolujte, zda konkrétní metadata a používat libovolně složité logické výrazy. Entrez umožňuje škálovat vyhledávání od základních až po pokročilé, jak si zúžit vyhledávání. Syntaxe pokročilých vyhledávání se může zdát skličující, ale SRA poskytuje grafické (https://www.ncbi.nlm.nih.gov/sra/advanced/) pro generování specifické syntaxe. A jak uvidíme níže, volič běhu Sra poskytuje ještě přátelštější uživatelské rozhraní pro zúžení vybraných dat.>> pohrajte si s SRA Entrez rozhraní, včetně advanced query builder, aby zjistili, zda můžete určit sadu SRA experimenty, které jsou relevantní pro jeden z vašich oblastí výzkumu.{: .hands_on}> # # # hands_on Hands-on: Generovat seznam odpovídajících experimentů pomocí Entrez>> Nyní, když máte základní znalost s SRA Entrez, pojďme najít sekvence použité v tomto kurzu.>> 1. Pokud tam ještě nejste, * * přejděte * * zpět na (https://www.ncbi.nlm.nih.gov/sra> 1. ** Vymazat * * jakýkoli vyhledávací text z vyhledávacího pole.> 1. ** Do vyhledávacího pole zadejte* * `sars-cov-2 ‚ a** klikněte * * `hledat`.> To vrátí delší seznam SRA experimenty, které odpovídaly naše vyhledávání, a že seznam je příliš dlouhý pro použití v tutoriálu cvičení. V tomto okamžiku bychom mohli použít pokročilý Tvůrce dotazů Entrez, o kterém jsme se dozvěděli výše.> Ale nebudeme. Místo toho umožňuje odeslat *příliš dlouhé pro tutorial* seznam výsledků, které máme k SRA Spustit Voliče, a používat jeho přátelštější rozhraní zúžit své výsledky.>> !(../../ obrázky / sra_entrez.GIF){: .hands_on}> # # # hands_on Hands-on: Jít z Entrez na SRA Spustit Voliče>> Zobrazit výsledky jako rozšířené interaktivní tabulky pomocí RunSelector.>> 1. Klepnutím na tlačítko Odeslat výsledky spusťte selektor, který se zobrazí v poli v horní části výsledků vyhledávání.>> !(../../obrázky / sra_entrez_result.png)>>> ### tip Co když nevidíte Spustit Volič Odkaz?>>>> možná Jste si všimli, tento text předtím, když jsi zkoumání Entrez vyhledávání. Tento text se zobrazí pouze po určitou dobu, když počet výsledků vyhledávání spadá do poměrně širokého okna. Neuvidíte to, pokud máte jen několik výsledků, a neuvidíte to, pokud máte více výsledků, než může selektor běhu přijmout.>>>> *musíte se dostat Spustit Volič pošlete své výsledky na Galaxy.* Co když nemáte dostatek výsledků pro spuštění tohoto odkazu? V takovém případě zavoláte na výběr spustit kliknutím * * * na rozbalovací nabídku „Odeslat do“ v pravém horním rohu panelu výsledků. Chcete-li se dostat ke spuštění selektoru, **vyberte** „Spustit selektor“ a poté **klepněte na tlačítko** na tlačítko „Jít“.> !(../../obrázky / sra_entrez_send_to.png)> {: .tip}>>> 1. ** Klikněte * * ‚Odeslat výsledky spustit selektor‘ v horní části panelu s výsledky vyhledávání. (Pokud tento odkaz nevidíte, podívejte se na komentář přímo výše.){: .hands_on}## Sra Run Selectordříve jsme se naučili, jak zúžit výsledky vyhledávání pomocí pokročilé syntaxe Entrez. Tuto sílu jsme však nevyužili, když jsme byli v Entrezu. Místo toho jsme použili jednoduché vyhledávání a poté jsme všechny výsledky odeslali do selektoru běhu. Zatím nemáme (krátký) seznam výsledků, na kterých bychom chtěli analýzu spustit. * Co to děláme?* Používáme Entrez a Run Selector, jak jsou navrženy pro použití: * pomocí rozhraní Entrez zúžit výsledky až na velikost, kterou volič Run může spotřebovat. * Odeslat tyto Entrez výsledky Sra Run Selector * použijte mnohem přátelštější rozhraní voliče běhu na 1. Snadněji pochopíme data, která máme 1. Zúžit tyto výsledky pomocí těchto znalostí.> ### komentář Spustit Volič je současně více i méně než Entrez>> Spustit Volič může dělat většina, ale ne všechny, co Entrez vyhledávání syntaxe může udělat. Run selector používá technologii * faceted search*, která se snadno používá a je výkonná, ale má vlastní limity. Konkrétně bude Entrez fungovat lépe při vyhledávání atributů, které mají desítky, stovky nebo tisíce různých hodnot. Run Selector bude fungovat lépe vyhledávání atributy s méně než 20 různých hodnot. Naštěstí to popisuje většinu vyhledávání.{: .komentář}okno výběru běhu je rozděleno do několika panelů:* *` ‚seznam filtrů’**: v levém horním rohu. To je místo, kde budeme upřesnit naše vyhledávání.* * * ` Select’**: souhrn toho, co bylo původně předáno Run Selector, a kolik z toho jsme dosud vybrali. (A zatím jsme nic z toho nevybrali.) Všimněte si také dráždivého, ale stále šedého tlačítka „Galaxy“.* * * ‚Found X Items‘ * * zpočátku se jedná o seznam položek odeslaných ke spuštění selektoru z Entrez. Tento seznam se zmenší, když na něj aplikujeme filtry.!(../../ obrázky / sra_run_selector.png)> # # # komentář proč se počet nalezených položek *zvýšil?*>> připomeňme, že rozhraní Entrez uvádí experimenty SRA (položky SRX). Run Selector lists * runs* – sekvenování datových sad – a tam jsou * jeden nebo více * běží na experiment. Máme stejná data jako dříve, nyní je vidíme jen v jemnějších detailech.{: .komentář} `Filtry Seznamu v levé horní části ukazuje sloupce v našich výsledcích, které mají buď souvislé číselné hodnoty, nebo 10 nebo méně (můžete změnit číslo) odlišné hodnoty v nich. ** Scroll * * dolů v seznamu vyberte několik filtrů. Když je vybrán filtr, zobrazí se níže pole * hodnoty*, seznam možností pro tento filtr a počet běhů s každou volbou. Tyto hodnoty / volby jsou vytaženy z metadat datové sady. Zkuste * * výběr * * několik zajímavých zvukových filtrů a poté * * vyberte * * jednu nebo více možností pro každý filtr. Zkuste * * zrušit výběr * * možností a filtrů. Jak to uděláte, počet nalezených výsledků se sníží nebo zvýší.> # # # tip Tip: Použijte Filtry k datům lépe porozumět>> Filtry jsou, jak si zmenšit datové soubory za úvahu pro odeslání do Galaxy, ale oni jsou také vynikající způsob, jak pochopit vaše data:> za Prvé, výběru filtru je snadný způsob, jak vidět rozsah hodnot ve sloupci. Možná nebudete moci (https://www.google.com/search?q=sra+sirs_outcome), ale můžete to zjistit tím, že uvidíte, jaké hodnoty jsou v něm.> za druhé můžete prozkoumat, jak se různé sloupce vzájemně vztahují. Existuje vztah mezi hodnotami ‚sirs_outcome‘ a` disease_stage‘?{: .tip}> ### hands_on Hands-on: Zúžit výsledky pomocí Run Voliče>> 1. Pokud máte zapnuté filtry, * * zrušte výběr * * je.> jakmile to uděláte, nebudou se pod seznamem filtrů zobrazovat žádné* hodnoty*.> 2. ** Zkopírujte a vložte * * tento vyhledávací řetězec do vyhledávacího pole „nalezené položky“.>> SRR11772204 NEBO SRR11597145 NEBO SRR11667145>> Tato ruka-vybral sadu běží omezuje naše výsledky do 3 běhy z různých geografických distribucí.{: .hands_on}tím se sníží seznam nalezených položek z desítek tisíc běhů na 3 běhy (zvládnutelné číslo pro tutoriál!). Ale ještě jsme s výběrem Run ještě neskončili. Všimněte si, že tlačítko „Galaxy“ je stále šedé. Zúžili jsme možnosti, ale ještě jsme nevybrali nic, co bychom mohli poslat do galaxie.Je možné vybrat každý zbývající běh * * kliknutím* * zaškrtnutí v horní části prvního sloupce. Můžete zrušit výběr vše * * kliknutím * * na `X`.> ### hands_on Hands-on: Zvolte běhy a poslat Galaxy>> 1. Vyberte všechny běží * * kliknutím * * na `X`.> a nyní je tlačítko `Galaxy` aktivní.> 1. ** Klikněte* * na tlačítko „Galaxy“ v sekci „Vybrat“ v horní části stránky.{: .hands_on} # # zpět v Galaxiikdyž klikneme na ‚Galaxy‘ V Run Selector, stane se několik věcí. Nejprve spustí novou kartu prohlížeče nebo okno, které se otevře v galaxii. Uvidíte * velký zelený rámeček*, který naznačuje, že handshake mezi SRA a Galaxy byl úspěšný, a poté uvidíte novou práci “ SRA “ ve vašem panelu Historie. Toto pole může začínat jako šedé / čekající, což znamená, že přenos ještě nezačal, nebo může jít přímo na žlutou / běžící nebo zelenou / hotovou.> # # # hands_on Hands-on: Zkontrolujte nový datový soubor Sra>> 1. Jakmile je přenos “ SRA “ dokončen, * * klikněte* * na ikonu galaxy-eye (eye) datové sady.>> zobrazuje datovou sadu v centrálním panelu Galaxy.{: .hands_on}datová sada “ SRA “ není sekvenční data, ale spíše * metadata*, která použijeme k získání sekvenčních dat ze SRA. Tato metadata odrážejí informace, které jsme viděli v sekci „nalezené položky“ v selektoru spuštění. Metadata nejsou koncová data, která hledáme od SRA, ale mít všechna tato metadata je často užitečné v následných krocích analýzy.Umožňuje nyní použít tato metadata k načtení sekvenčních dat ze SRA. SRA poskytuje nástroje pro extrahování všech druhů informací, včetně samotných sekvenčních dat. Nástroj Galaxy „rychlejší stahování a extrahování čte v FASTQ“ je založen na nástroji SRA (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump) a dělá to právě.–>
- Najít potřebné údaje v SRA
- hands_on Hands-on: Úkol popis
- komentář Komentář
- Process and filter SraRunInfo.soubor csv v galaxii
- hands_on Hands-on: Upload SraRunInfo.csv soubor do Galaxie
- komentář pozor na řezy
- hands_on Hands-on: Vytvoření podmnožiny dat
- tip Tip: Hledání nástrojů
- ke Stažení sekvenačních dat s Rychlejší Stahování a Extrakt Čte v FASTQ
- hands_on Hands-on: Úkol popis
- co teď?
- variační analýza sekvenačních dat SARS-Cov-2
- komentujte použitígalaxy.* Projekt analýzy COVID-19
- Získejte referenční genom data
- hands_on Hands-on: ten referenční genom údaje
- Tip: Import přes odkazy
- Adaptér ořezávání s fastp
- hands_on Hands-on: Úkol popis
- Zarovnání s Mapou s BWA-MEM
- hands_on Hands-on: Zarovnání sekvenčních čtení referenční genom
- Odebrat duplicity s MarkDuplicates
- hands_on Hands-on: odstranit PCR duplikáty
- Vytvořit zarovnání statistiky s Samtools statistiky
- hands_on Hands-on: Vytvářet zarovnání statistiky
- Zarovnání čte s lofreq viterbiho
- hands_on Hands-on: Přestavět čte kolem indels
- Přidat indel vlastnosti s lofreq-li Vložit indel vlastnosti
- hands_on Hands-on: Přidat indel vlastnosti
- Volání Variant pomocí lofreq Zavolat varianty
- hands_on Hands-on: Call varianty
- Anotujte variantní efekty pomocí SnpEff eff:
- hands_on Hands-on: Komentovat varianta účinky
- vytvořte tabulku variant pomocí Snpsift Extract Fields
- hands_on Hands-on: Vytvořit tabulku variant
- shrňte data pomocí MultiQC
- hands_on Hands-on: Shrnout údaje
- Závěr
- keypoints Klíčové body
- Nejčastější Otázky
- Užitečné literatura
- zpětná vazba
- Odvoláním na tomto Tutoriálu
- details BibTeX
Najít potřebné údaje v SRA
Nejprve musíme najít dobré dataset hrát. Sequence Read Archive (Sra) je primární archiv nesestavených čtení provozovaný americkým Národním institutem zdraví (NIH). SRA je skvělým místem pro získání sekvenčních dat, která jsou základem publikací a studií. Pojďme to udělat:
hands_on Hands-on: Úkol popis
- Přejít na NCBI je SRA stránce tím, že poukáže prohlížeče https://www.ncbi.nlm.nih.gov/sra
- Do pole hledat zadejte
SARS-CoV-2 Patient Sequencing From Partners / MGH(Alternativně, můžete jednoduše klikněte na tento odkaz)
- webová stránka bude zobrazovat velké množství SRA datových souborů (v době psaní tohoto článku tam byly 2,223). Toto jsou data ze studie popisující analýzu SARS-CoV-2 v oblasti Bostonu.
- ke Stažení metadat popisujících datové soubory:
- kliknutí na Odeslat: dropdown
- Výběr
File- Změna Formátu
RunInfo- Klepnutím na tlačítko Vytvořit fileHere je, jak by to mělo vypadat:
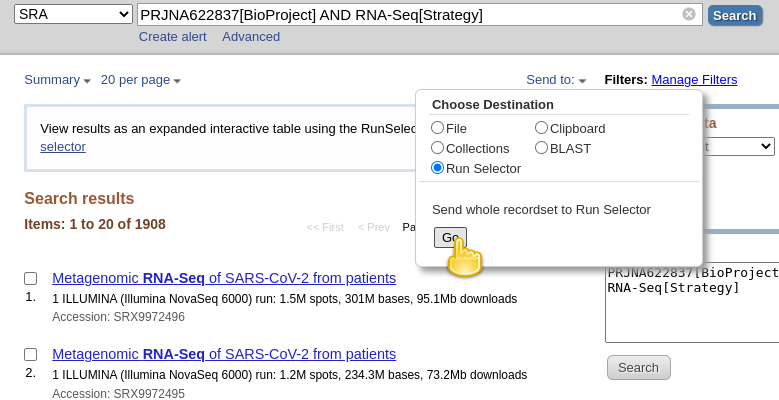
- To by se vytvořit poměrně velké
SraRunInfo.csvsouborDownloadssložka.
Nyní, když jsme tento soubor stáhli, můžeme přejít do instance Galaxy a začít jej zpracovávat.
komentář Komentář
Všimněte si, že soubor, který jsme právě stáhli, není sekvenčních dat sám. Spíše jde o metadata popisující vlastnosti sekvenování čtení. Tento seznam filtrujeme na několik přístupů, které budou použity ve zbývající části tohoto tutoriálu.
Process and filter SraRunInfo.soubor csv v galaxii
hands_on Hands-on: Upload SraRunInfo.csv soubor do Galaxie
- Přejít na Galaxy instance volba, jako jeden z usegalaxy.org, usegalaxy.eu, usegalaxy.org.au nebo jakýkoli jiný. (Tento návod používá usegalaxy.org).
- klikněte na tlačítko Nahrát Data:
- V dialogovém okně, které se objeví klikněte na „Vybrat místní soubory“ tlačítko:

- vyhledejte a vyberte
SraRunInfo.csvsoubor z vašeho počítače- Klepněte na tlačítko Start
- Zavřete dialogové okno stisknutím tlačítka Zavřít
- nyní můžete podívat na obsah tohoto souboru kliknutím galaxy-oka (očí) ikonu. Uvidíte, že tento soubor obsahuje mnoho informací o jednotlivých přístupech SRA. V této studii každý přístup odpovídá jednotlivému pacientovi, jehož vzorky byly sekvenovány.
Galaxy může zpracovat všechny 2,000+ datové soubory, ale aby se tento kurz snesitelné musíme vybraných menší podskupiny. Zejména naše předchozí zkušenosti s těmito daty ukazují dva zajímavé datové soubory SRR11954102 a SRR12733957. Tak je vytáhneme.
komentář pozor na řezy
níže uvedená praktická část používá řezný nástroj. V galaxii jsou z historických důvodů dva řezané nástroje. Tento příklad používá nástroj s celým názvem vyjmout sloupce z tabulky (vyjmout). Stejná logika však platí i pro druhý nástroj. Má prostě trochu jiné rozhraní.
hands_on Hands-on: Vytvoření podmnožiny dat
- Najít nástroj „Vyberte řádky, které odpovídají výrazu“ nástroj v Filtrovat a Třídit části panelu nástrojů.
tip Tip: Hledání nástrojů
Galaxy může mít ohromující množství nástrojů nainstalován. Chcete-li najít konkrétní typ nástroje, vyhledejte nástroj ve vyhledávacím poli panel nástrojů.
- ujistěte Se, že
SraRunInfo.csvdataset právě jsme nahráli je uveden v param-file „Vyberte řádky z“ pole formuláře nástroj.- do pole“ vzor „zadejte následující výraz →
SRR12733957|SRR11954102. Jedná se o dva přístupy, které chceme najít oddělené symbolem potrubí||or: najděte řádky obsahujícíSRR12733957neboSRR11954102.- klikněte na tlačítko
Execute.- tím se vygeneruje soubor obsahující dva řádky (dobře … jeden řádek se také používá jako záhlaví, takže se objeví soubor má tři řádky. To je v pořádku.)
- vyjměte první sloupec ze souboru pomocí nástroje „vyjmout“ nástroj, který najdete v části manipulace s textem v podokně nástroje.
- ujistěte se, že datová sada vytvořená předchozím krokem je vybrána v poli „Soubor k vyjmutí“ formuláře nástroje.
- změňte „oddělený“ na
Comma- v“ seznamu polí „vyberte
Column: 1.- Hit
Executevytvoří textový soubor s jen dva řádky:SRR12733957SRR11954102
Nyní, že máme identifikátory souborů dat, které chceme, musíme stáhnout aktuální údaje o sekvenování.
ke Stažení sekvenačních dat s Rychlejší Stahování a Extrakt Čte v FASTQ
hands_on Hands-on: Úkol popis
- Rychlejší Stáhnout a Extrahovat Čte v FASTQ nástroj s následujícími parametry:
- „vyberte typ vstupu“:
List of SRA accession, one per line
- parametr param-soubor „sra přistoupení seznam“ by měl bod výstupu nástroje „Cut“ z předchozího kroku.
- klikněte na tlačítko
Execute. Tím se spustí nástroj, který načte datové sady pro čtení sekvence pro běhy, které byly uvedeny v datovém souboruSRA. Může to nějakou dobu trvat. Takže to může být dobrý čas na kávu.- při odeslání této úlohy se v panelu Historie vytvoří několik záznamů:
Pair-end data (fasterq-dump): Obsahuje Spárované-end datové sady (pokud je k dispozici)Single-end data (fasterq-dump)Obsahuje Single-end datové sady (pokud je k dispozici)Other data (fasterq-dump)Obsahuje Nepárové datové sady (pokud je k dispozici)fasterq-dump logObsahuje Informace o nástroj realizace
první tři položky jsou ve skutečnosti kolekce datových souborů. Kolekce v galaxii jsou logické seskupení datových souborů, které odrážejí sémantické vztahy mezi nimi v experimentu / analýze. V tomto případě nástroj vytvoří samostatnou sbírku pro párová čtení, jednotlivá čtení a další.Další informace naleznete v návodech ke sbírkám.
Prozkoumejte sbírky tak, že nejprve kliknete na název kolekce v panelu Historie. To vás zavede do sbírky a ukáže vám datové sady v ní. Poté můžete přejít zpět na vnější úroveň své historie.
jakmile fasterq dokončí přenos dat (všechny boxy jsou zelené / hotovo), jsme připraveni je analyzovat.
co teď?
nyní můžete analyzovat získaná data pomocí jakýchkoli nástrojů pro analýzu sekvencí a pracovních postupů v galaxii. SRA uchovává podkladová data pro každý představitelný typ experimentu *-seq.
Pokud jste spustili tento tutoriál, ale načtené datové sady, které vás zajímaly, podívejte se na zbytek knihovny GTN, kde najdete nápady, jak analyzovat v galaxii.
Pokud jste však načetli datové sady použité v příkladech tohoto tutoriálu výše, jste připraveni spustit analýzu variant SARS-CoV-2 níže.
variační analýza sekvenačních dat SARS-Cov-2
v této části tutoriálu provedeme variantní volání a základní analýzu výše stažených datových sad. Začneme stažením Wuhan-Hu-1 SARS-CoV-2 referenční sekvence, pak spustit adaptér oříznutí, vyrovnání a varianta volání a nakonec se podíváme na geografické rozložení některých z nalezených variant.
komentujte použitígalaxy.* Projekt analýzy COVID-19
Tento tutoriál používá podmnožinu dat a prochází analýzourozlišování covid19.galaxyproject.organizace.Údaje pro covid19.galaxyproject.org isbeing průběžně aktualizován jako nové datové sady jsou zveřejněny.
Získejte referenční genom data
referenční genom dat je dnes pro SARS-CoV-2, „těžký akutní respirační syndrom koronavirus 2 izolovat Wuhan-Hu-1, kompletní genom“, které mají přistoupení ID NC_045512.2.
tato data jsou k dispozici od Zenodo pomocí následujícího odkazu.
hands_on Hands-on: ten referenční genom údaje
Importovat následující soubor do své historie:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzTip: Import přes odkazy
- Kopírovat umístění odkazu
- Otevřete Galaxy Upload Manager (galaxy-nahrát na pravé horní části panelu nástrojů)
- Zvolte Vložit/Načíst Data
- Vložit odkaz do textového pole
- Stiskněte tlačítko Start
- Zavřít windowBy default, Galaxy používá URL jako název, tak přejmenovat soubory, s více užitečných jméno.
Adaptér ořezávání s fastp
Odstranění sekvencování adaptérů zlepšuje zarovnání a varianta volání. fastp nástroj může automaticky detekovat široce používané sekvenční adaptéry.
hands_on Hands-on: Úkol popis
- fastp nástroj s následujícími parametry:
- „Single-end, nebo spárované čte“:
Paired Collection
- param-file „Zvolte spárované kolekce(s)“:
list_paired(výstup Rychleji Stáhnout a Extrahovat Čte v FASTQ nástroj)- V „Možnosti Výstupu“:
- „Výstup JSON report“:
Yes
Zarovnání s Mapou s BWA-MEM
BWA-MEM nástroj je široce používán sekvence vyrovnávač pro krátké sekvenční čtení datových souborů, jako jsou ty, analyzujeme v tomto návodu.
hands_on Hands-on: Zarovnání sekvenčních čtení referenční genom
- Mapa s BWA-MEM nástroj s následujícími parametry:
- „vyberte referenční genom z historie nebo použít vestavěný index?“:
Use a genome from history and build index
- param-soubor „Použít následující dataset jako referenční sekvence“:
output(Input dataset)- „Jeden nebo Spárované-end čte“:
Paired Collection
- param-file „Zvolte spárovaný kolekce“:
output_paired_coll(výstup fastp nástroj)- „Nastavit číst skupin informací?“:
Do not set- „Vyberte režim analýzy“:
1.Simple Illumina mode
Odebrat duplicity s MarkDuplicates
MarkDuplicates nástroj odstraňuje duplicitní sekvence pocházející z knihovny příprava artefakty a sekvenování artefakty. Je důležité odstranit tyto artefakty sekvence, aby se zabránilo umělé nadměrné reprezentaci jedné molekuly.
hands_on Hands-on: odstranit PCR duplikáty
- Markduplikáty nástroj s následujícími parametry:
- param-file „Vyberte SAM/BAM datového souboru nebo datové sady kolekce“:
bam_output(výstup Mapa s BWA-MEM nástroj)- „Pokud je to pravda, nepište duplikáty do výstupního souboru místo psaní s příslušnou vlajky set“:
Yes
Vytvořit zarovnání statistiky s Samtools statistiky
Poté, co duplicitní značení krok výše uvedeného lze generovat statistiky o vyrovnání jsme si vytvořili.
hands_on Hands-on: Vytvářet zarovnání statistiky
- Samtools statistiky nástroj s následujícími parametry:
- param-soubor „BAM“:
outFile(výstup MarkDuplicates nástroj)- „Nastavit pokrytí distribuce“:
No- „Výstup“:
One single summary file- „Filtrovat podle SAM vlajky“:
Do not filter- „Použít referenční sekvence“:
No- „Filtr podle krajů“:
No
Zarovnání čte s lofreq viterbiho
Přestavět čte nástroj opravuje výchylkami kolem inserce a delece. To je nutné pro přesné zjištění variant.
hands_on Hands-on: Přestavět čte kolem indels
- Zarovnání čte s lofreq nástroj s následujícími parametry:
- param-soubor „, – uvádí se vazba“:
outFile(výstup MarkDuplicates nástroj)- „Vybrat zdroj pro referenční genom“:
History
- param-soubor „Reference“:
output(Input dataset)- V „Advanced options“:
- „Jak zvládnout základní vlastnosti 2?“:
Keep unchanged
Přidat indel vlastnosti s lofreq-li Vložit indel vlastnosti
Tento krok přidá indel vlastnosti do našeho zarovnání souborů. To je nezbytné pro volání variant pomocí variant volání pomocí nástroje lofreq
hands_on Hands-on: Přidat indel vlastnosti
- Vložit indel vlastnosti s lofreq nástroj s následujícími parametry:
- param-soubor „Čte“:
realigned(výstup Přestavět čte nástroj)- „Indel výpočtu přístup“:
Dindel
- „Vybrat zdroj pro referenční genom“:
History
- param-soubor „Reference“:
output(Vstupní datový soubor)
Volání Variant pomocí lofreq Zavolat varianty
nyní Jsme připraveni zavolat variant.
hands_on Hands-on: Call varianty
- Volání varianty s lofreq nástroj s následujícími parametry:
- param-soubor „Vstupních čte ve formátu BAM“:
output(výstup Vložit indel vlastnosti nástroj)- „Vybrat zdroj pro referenční genom“:
History
- param-soubor „Reference“:
output(Input dataset)- „Hovor variant napříč“:
Whole reference- „Typy variant zavolat“:
SNVs and indels- „Varianta volání parametry“:
Configure settings
- V „Krytí“:
- „Minimální pokrytí“:
50- V „Base calling“:
- „Minimální baseq“:
30- „Minimální baseq pro alternativní základny“:
30- v „mapování qualityy
20- „varianta parametry filtru“:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
výstup z tohoto kroku je sbírka VCF soubory, které lze vizualizovat v genomu prohlížeče.
Anotujte variantní efekty pomocí SnpEff eff:
nyní anotujeme varianty, které jsme nazvali v předchozím kroku, s účinkem, který mají na genom SARS-CoV-2.
hands_on Hands-on: Komentovat varianta účinky
- SnpEff eff: nástroj s následujícími parametry:
- param-soubor „Sekvence změny (Snp, MNPs, InDels)“:
variants(výstup z Volání varianty nástroj)- „; Výstupní formát“:
VCF (only if input is VCF)- „Vytvořit zprávy ve formátu CSV, což je užitečné pro následné analýzy (-csvStats)“:
Yes- „Anotace options“: `
- „výstup Filtru“: `
- „odfiltrovat konkrétní Účinky“:
No
výstup z tohoto kroku je soubor VCF s přidanou varianta účinky.
vytvořte tabulku variant pomocí Snpsift Extract Fields
nyní vybereme různé efekty z VCF a vytvoříme tabulkový soubor, který je pro člověka srozumitelnější.
hands_on Hands-on: Vytvořit tabulku variant
- SnpSift Extrakt Pole nástroj s následujícími parametry:
- param-soubor „Varianta vstupního souboru ve formátu VCF“:
snpeff_output(výstup SnpEff eff: nástroj)- „Pole extrahovat“:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- „multiple field separator“:
,- „prázdné pole text“:
.
zkontrolujte, zda není výstupní soubory a uvidíme, zkontrolujte, zda Variant v tomto souboru jsou také popsány v pozorovatelný notebook, který ukazuje geografické rozložení SARS-CoV-2 varianta sekvence,
Zajímavé varianty zahrnují C k T varianta na pozici 14408 (14408C/T) v SRR11772204, 28144T/C v SRR11597145 a 25563G/T v SRR11667145.
shrňte data pomocí MultiQC
nyní shrneme naši analýzu pomocí MultiQC, která generuje krásnou zprávu pro naše data.
hands_on Hands-on: Shrnout údaje
- MultiQC nástroj s následujícími parametry:
- V „Výsledky“:
- param-opakovat „Vložit Výsledky,“
- „Který nástroj byl použit generovat protokoly?“:
fastp
- param-file „výstup fastp“:
report_json(výstup fastp nástroj)- param-opakovat „Vložit Výsledky,“
- „Který nástroj byl použit generovat protokoly?“:
Samtools
- V „Samtools výstup“:
- param-opakovat „Vložit Samtools výstup“
- „Typ Samtools výstup?“:
stats
- param-file“Samtools stats output“:
output(výstup z Samtools statistiky nástroj)- param-opakovat „Vložit Výsledky,“
- „Který nástroj byl použit generovat protokoly?“:
Picard
- v „Picard output“:
- param-repeat „Insert Picard output“
- “ typ výstupu Picard?“:
Markdups- param-file „Picard output“:
metrics_file(výstup MarkDuplicates nástroj)- param-opakovat „Vložit Výsledky,“
- „Který nástroj byl použit generovat protokoly?“:
SnpEff
- param-soubor „Výstup SnpEff“:
csvFile(výstup SnpEff eff: nástroj)
Závěr
Gratulujeme, teď víte, jak importovat sekvence dat z SRA a jak spustit příklad analýzy v těchto datových souborů.
keypoints Klíčové body
Pořadí údajů v SRA mohou být přímo importovány do Galaxie.
Nejčastější Otázky
Máte otázky o tomto kurzu? Podívejte se na stránku FAQ pro téma analýzy Variant a zjistěte, zda je zde uvedena vaše otázka. Pokud ne, prosím, zeptejte se na vaši otázku na GTN Gitter Kanálu nebo na Galaxy Fóru Nápovědy
Užitečné literatura
Další informace, včetně odkazů na dokumentaci a originální publikací, pokud jde o nástroje, techniky analýzy a interpretace výsledků popsaných v tomto kurzu naleznete zde.
zpětná vazba
Použili jste tento materiál jako instruktor? Neváhejte a dejte nám zpětnou vazbu o tom, jak to šlo.
Odvoláním na tomto Tutoriálu
- Marius van den Beeka, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 Z NCBI je Sekvence Číst Archiv (SRA) pro Galaxy: SARS-CoV-2 varianta analýzy (Galaxy Školicí Materiály). / školení-materiál / témata/varianta-analýza / návody / sars-cov-2 / výukový program.html Online; přístupné dnes
- Batut et al., 2018 komunitní trénink analýzy dat pro biologické buněčné systémy 10.1016 / j. cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}