aminokyseliny, nukleotidy nebo jakýkoli jiný evoluční charakter jsou v určité míře nahrazeny jinými. Představte si například, evoluční sekvence s threepossible státy, A, B a C. Pokud substituční model je časově reverzibilní, therewill být tři přechodu sazby,<>B, B<>C, a<>C.
Předpokládejme, že sazby jsou 1, 1 a 0 v jednotkách substituce na 100znak za jednotku času. Po jedné jednotky času, ve 300 znaků longsequence původně složená rovnoměrně z As, Bs a Cs, očekáváme, že tam byly jednou A B náhrada a B C substituce. Pokud porovnáváme dvě homologní sekvence v živých organismech, protože jedna jednotka času prošla pro obě sekvence, očekávali bychom dvě a až B a dvě B až Csubstituce mezi současnými sekvencemi.
Bez ohledu na to, jak dlouho jsme se spustit tento proces, tam bude nikdy být directreplacement z C. k Dispozici bude také být nikdy A až C substituce v rámci aso-tzv. nekonečný míst modelu, kde je více než jedna substituce může occurat jednom místě.
Nicméně od A do B a z B do C substituce jsou společné, v rámci konečných sitesmodel nakonec B bude nahrazen C, v místě, kde byl previouslyreplaced B. Tato nepřímá náhrada C (nebo ekvivalentně v atime-reverzibilní model, C) stává spíše delší době periodseparating na homologních sekvencí.
simuloval jsem vývoj sekvencí na základě výše uvedeného scénáře, který prováděl thesimulaci po dobu 10 jednotek času. Z této substituce jsem pozoroval počty následků pro každý vzor webu:
| A | B | C | |
|---|---|---|---|
| A | 91 | 9 | 0 |
| B | 5 | 86 | 9 |
| C | 0 | 9 | 91 |
Během této relativně krátké trvání, nezdá se, jako kdyby všechny<>Csubstitutions došlo. Nicméně, když jsem reran simulaci pro 100 unitsof času:
| A | B | C | |
|---|---|---|---|
| A | 55 | 35 | 10 |
| B | 29 | 36 | 35 |
| C | 20 | 36 | 44 |
Jak můžete vidět, mnoho „A“ znaky byly nahrazeny „C“ a naopak. Více obecně, v rámci konečných lokalit model více substitutionscause rozložení stránky vzor počítá, aby se stal mnohem plošší beyondsimply zvyšující se podíl mimo-diagonální vzhledem k úhlopříčce se počítá.Matrice skóre Pam a BLOSUM představují více substitucí různými způsoby.
PAM matice pro aminokyseliny, spolu s jedním písmenem abbreviationsused pro geneticky kódované aminokyseliny, byly vyvinuty MargaretDayhoff. Byly původně publikovány v roce 1978, a na základě proteinsekvences Dayhoff byl sestavování od roku 1960, publikoval jako theAtlas proteinové sekvence a struktury.
název PAM pochází z „point accepted mutation“ a odkazuje na nahrazení jedné aminokyseliny v proteinu jinou aminokyselinou.Tyto mutace byly identifikovány porovnáním velmi podobné sekvence s alespoň 85% identity, a předpokládá se, že veškeré pozorované substituce byly vsledkem jedné mutace mezi rodové posloupnosti a jeden z přítomností den sekvence.
PAM také definuje časovou jednotku, kde 1 PAM je čas, ve kterém se očekává, že 1/100 aminokyselin podstoupí mutaci. Na PAM1 pravděpodobnostní matice ukazují, že pravděpodobnost, že se aminokyseliny ve sloupci j je nahrazena aminokyselina na řadě já. To byla vypočítána z Dayhoff je PAM počítá, a schopnosti tobe 1 PAM jednotku času. Jak můžete vidět, mimo-diagonální pravděpodobnosti v thePAM1 matice jsou velmi malé (všechny prvky byly měřítko 10 000 forlegibility):
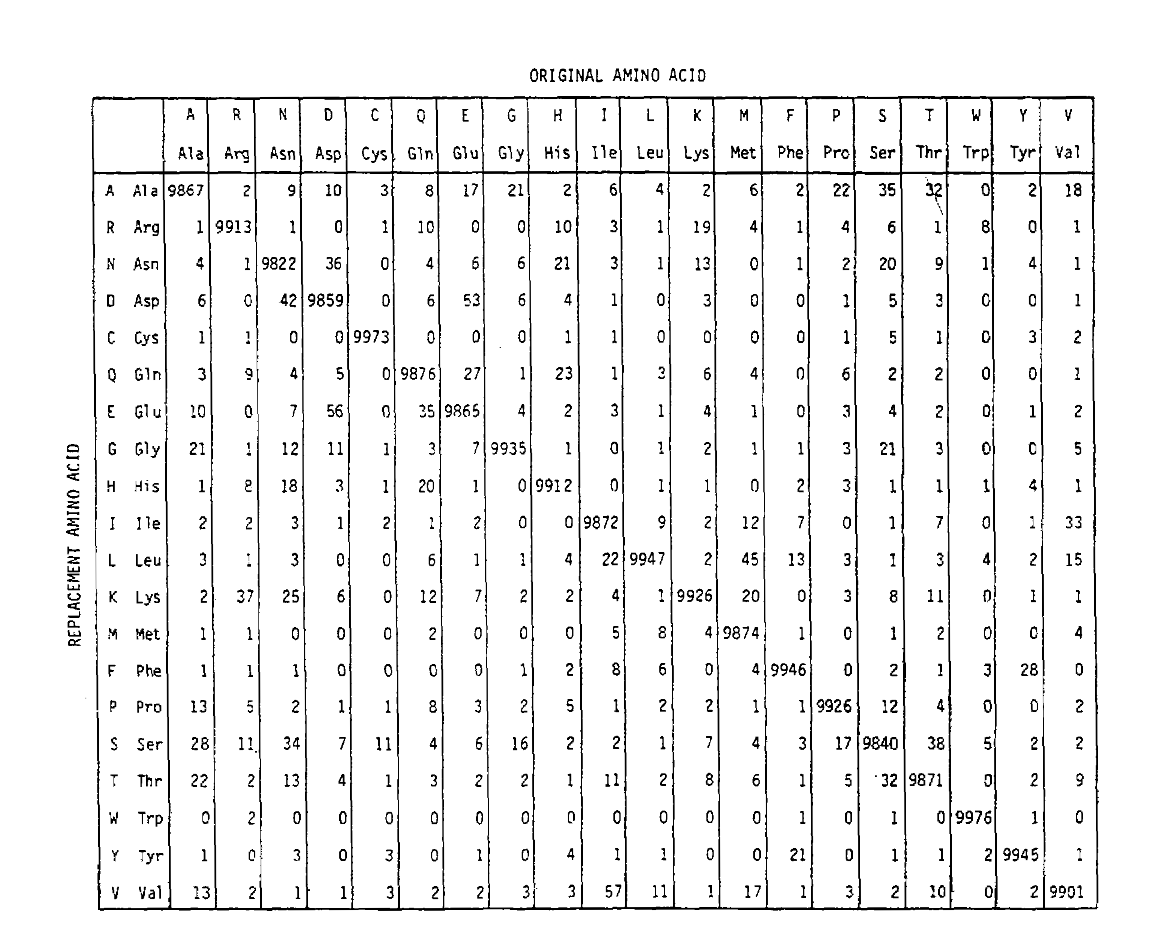
vypočítat aminokyselin náhradní pravděpodobnosti pro delší timedurations, matice lze násobit sama o sobě correspondingnumber časy. Tak PAM250 pravděpodobnostní matice, popisující thereplacement pravděpodobnosti vzhledem 250 PAM jednotky času, byl odvozen od zvedání hodnoty pravděpodobnostní matice PAM1 do výkonu 250 (všechny prvky byly odstupňovány podle 100for čitelnost):
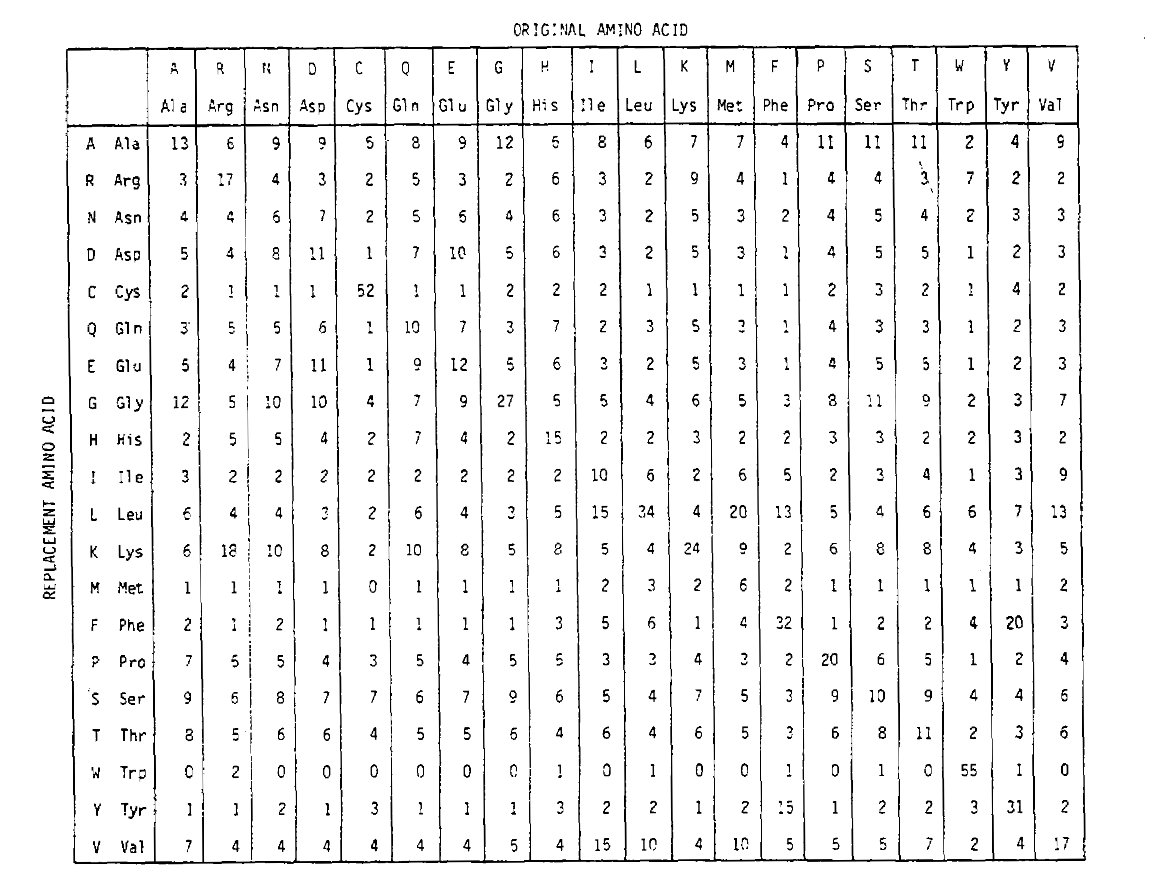
náhradní pravděpodobnosti odvozené pomocí tohoto umocňování correctlyaccount pro vícenásobné substituce. Nejen, že jsou off-diagonalprobabilities proporcionálně větší, jak byste očekávali na delší timeduration, ale jsou plošší. Například, pravděpodobnost, valin (V)isoleucin (I) výměna je 33× větší než V histadine (H)výměna v matice PAM1, ale pouze 4,5× větší v matice PAM250.
matrice skóre pak lze vypočítat z matic pravděpodobnosti a základních frekvencí.
matice BLOSUM, vyvinuté Stevenem a Jorjou Henikoffovými a publikované v1992, má velmi odlišný přístup. Vzhledem k tomu, že PAM je implicitně applyinga stacionární konečných lokalit model evoluce pomocí maticové umocňování, účinky více substituce je řešena implicitně v BLOSUM byconstructing různé skóre matice na různých časových měřítcích.
v rámci více sekvenčních zarovnání homologních sekvencí jsou identifikovány konzervované sousedící bloky aminokyselin. V rámci každého bloku, multiplesekvence jsou seskupeny, když jejich párová průměrná identita sekvence je vyšší než nějaký práh. Prahová hodnota je 80% pro matici BLOSUM80, 62% pro BLOSUM62, 50% pro BLOSUM50 a tak dále.
to znamená, že pro BLOSUM80 budou mít bloky průměrnou párovou identitu větší než 80%, pro BLOSUM62 ne větší než 62% atd.
pravděpodobnosti náhrady aminokyselin pro homologní sekvence jsou vypočteny z párových srovnání mezi klastry. Tyto pravděpodobnosti budou výsledkem jedné a více substitucí, s vícenásobnými substitucemi, které mají větší vliv na větší evoluční vzdálenosti. Proto scorematrices generovány z párového srovnání mezi shluky na averagegreater vzdálenost, jako BLOSUM50 matrix, bude přirozeně účet pro thelarger efekt vícenásobné substituce.
ačkoli se vydávají různými cestami, konečné skóre blosum a PAM jsou ve skutečnosti docela podobné. Podle Henikoffa a Henikoffa jsou následující matrice Pam a BLOSUM srovnatelné:
| PAM | BLOSUM |
|---|---|
| PAM250 | BLOSUM45 |
| PAM160 | BLOSUM62 |
| PAM120 | BLOSUM80 |
For more information on PAM (Dayhoff) and BLOSUM matrices, see kapitola 2biologická sekvenční analýza Durbin et al., a Wikipedia.
Aktualizace 13. října 2019: pro další pohled na substituční matice, obraťte se na „Objížďky“ oddíl na konci Kapitoly 5 Bioinformatických Algoritmů (2nd nebo 3rd Edition) Compeau a Pevzner.