tämän opetusohjelman tavoitteena on esitellä sinut seuraavan sukupolven sekvensointitietojen käsittelyyn Galaxyssa. Tämä opetusohjelma käyttää COVID-19-muunnosta, joka kutsuu Illumina-datasta, mutta kyse ei ole muunnospuhelusta sinänsä.
tämän opetusohjelman valmistuttua tiedät:
- Miten löytää dataa SRA: sta ja siirtää tämä tieto Galaxyyn
- miten suorittaa PERUSNG-tietojenkäsittely Galaxyssa mukaan lukien:
- Illumina-tietojen laadunvalvonta (QC)
- kartoitus
- kaksoiskappaleiden poistaminen
- variantti kutsuu
lofreq
- variantti-merkintä
- käyttäen tietoaineistokokoelmia
- tuo tietoa Jupyteriin
# # Agenda>> tässä tutoriaalissa käydään läpi:>> 1. TOC> {:toc}>{: .agenda} # # kaksi polkua tämän tutoriaalin läpi loimme kaksi liikerataa, joita voit seurata tämän opetusohjelman kautta.1. ** Liikerata 1 * * – Aloita NCBI: n SRA: sta ja etsi käytettävissä olevia liitteitä → Aloita (#the-sequence-read-archive)2. ** Liikerata 2* * – ohittaa NCBI: n SRA ja aloittaa Galaxy suoraan. → Käynnistä (#back-in-galaxy)suosittelemme aloittamaan * * liikerata 2**.# Sekvenssin Lukuarkisto (https://www.ncbi.nlm.nih.gov/sra) on *kokoamattomien lukujen* ensisijainen arkisto (https://www.ncbi.nlm.nih.gov/). SRA on hyvä paikka saada sekvensointitietoja, jotka ovat julkaisujen ja tutkimusten taustalla.Tämä opetusohjelma kattaa miten saada sekvenssitiedot Sra osaksi Galaxy käyttämällä suoraa yhteyttä kahden.> # # kommentti>> kuulet Sra: sta myös nimityksen *Short Read Archive*, sen alkuperäinen nimi.> {:.kommentti}## pääsy SRASRA pääsee joko suoraan sen verkkosivuilla, tai työkalupaneelin Galaxy.> # # comment kommentoi>> aluksi työkalupaneeli optio Sra: n käyttöön on olemassa vain (https://usegalaxy.org/). Tuki suora yhteys SRA sisältyy 20.05 julkaisu Galaxy {:.kommentti}> # # hands_on Hands-on: Explore Sra Entrez>> 1. Mene galaksisi valikoimaan, kuten johonkin (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) tai mihin tahansa muuhun. (Tämä opetusohjelma käyttää usegalaxy.org).> 1. Jos historiasi ei ole jo tyhjä, Aloita uusi historia (Katso (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) lisää Galaksihistorioista)> 1. ** Klikkaa * *` Get Data ’ yläreunassa työkalupaneelin.> 1. ** Napsauta * * ’Sra Server’ Työkaluluettelossa ’Get Data’.> tämä vie (https://www.ncbi.nlm.nih.gov/sra) — voit aloittaa myös suoraan SRA: sta. Hakukenttä näkyy sivun yläosassa. Kokeile etsiä jotain olet kiinnostunut, kuten `delfiini` tai `munuainen` tai `delfiini munuainen` ja sitten **klikkaa** `Haku` painiketta.>> tämä palauttaa luettelon *SRA-kokeista*, jotka vastaavat hakusanaa. SRA-kokeet, jotka tunnetaan myös nimellä * SRX-merkinnät*, sisältävät sekvenssitiedot tietystä kokeesta sekä selityksen itse kokeesta ja muista siihen liittyvistä tiedoista. Voit tutustua palautettuihin kokeisiin klikkaamalla niiden nimeä. Katso lisää (https://www.ncbi.nlm.nih.gov/books/NBK56913/) (https://www.ncbi.nlm.nih.gov/books/n/helpsrakb/).>> kun syötät tekstiä SRA-hakukenttään, käytät (https://www.ncbi.nlm.nih.gov/sra/docs/srasearch/). Entrez tukee sekä yksinkertaisia tekstihakuja että hyvin tarkkoja hakuja, joissa tarkistetaan tiettyjä metatietoja ja käytetään mielivaltaisen monimutkaisia loogisia ilmaisuja. Entrez avulla voit skaalata haut perus-ja advanced kun kavennat hakuja. Tarkennettujen hakujen syntaksi voi tuntua pelottavalta, mutta SRA tarjoaa graafisen (https://www.ncbi.nlm.nih.gov/sra/advanced/) tietyn syntaksin luomiseksi. Ja, kuten näemme alla, SRA Run Selector tarjoaa vieläkin ystävällisempi käyttöliittymä kaventaa valittujen tietojen.>> leikittele SRA Entrez-käyttöliittymällä, mukaan lukien advanced query builder, nähdäksesi, pystytkö tunnistamaan joukon Sra-kokeita, jotka ovat merkityksellisiä jollekin tutkimusalueellesi.{: .hands_on}> # # # hands_on Hands-on: Luo lista vastaavista kokeista käyttäen Entrez>> nyt kun sinulla on perustiedot SRA Entrezistä, etsitään tässä tutoriaalissa käytetyt sekvenssit.>> 1. Jos et ole jo siellä, * * navigoi * * takaisin (https://www.ncbi.nlm.nih.gov/sra> 1. ** Tyhjennä* * kaikki hakutekstit hakukentästä.> 1. ** Kirjoita hakukenttään **` sars-cov-2` ja **klikkaa** `Haku`.> tämä palauttaa pitkän listan Sra-kokeista, jotka vastaavat meidän hakuamme, ja tämä lista on aivan liian pitkä käytettäväksi opetusharjoituksessa. Tässä vaiheessa voisimme käyttää advanced Entrez kysely rakentaja opimme edellä.> mutta emme. sen sijaan lähetetään *liian pitkä opetusohjelma* lista tuloksia meillä on SRA Run-valitsimeen, ja käytetään sen ystävällisempää käyttöliittymää rajaamaan tuloksiamme.>> !(../../ images / sra_entrez.png) {: .hands_on}> # # # hands_on Hands-on: Siirry Entrezistä Sra: n Juoksuvalitsimeen>> Katso tulokset laajennettuna interaktiivisena taulukkona käyttäen Juoksuvalitsinta.>> 1. Napsauta Lähetä tulokset ajaaksesi valitsimen, joka ilmestyy hakutulosten yläreunassa olevaan ruutuun.>> !(../../ images / sra_entrez_result.png)>>> # # vinkki mitä jos et näe ajetun valitsimen linkkiä?>>>> olet saattanut huomata tämän tekstin aiemmin tutkiessasi Entrez-hakua. Tämä teksti näkyy vain osan ajasta, kun hakutulosten määrä osuu melko laajaan ikkunaan. Et näe sitä, jos sinulla on vain muutamia tuloksia, ja et näe sitä, jos sinulla on enemmän tuloksia kuin ajaa valitsin voi hyväksyä.>>>>* Entä jos sinulla ei ole tarpeeksi tuloksia käynnistääksesi tämän linkin näyttämisen? Siinä tapauksessa soitat get to the Run-valitsimeen** klikkaamalla * * `Send to` pulldown-valikkoa tulospaneelin oikeassa yläkulmassa. Pääset suorittamaan valitsimen,** valitse **`Suorita Valitsin` ja sitten** klikkaa * * `mene` painiketta.> !(../../ images / sra_entrez_send_to.png)> {: .kärki}>>> 1. ** Napsauta * * ’Lähetä tulokset Suorita valitsin’ hakutulospaneelin yläreunassa. (Jos et näe tätä linkkiä, niin katso kommentti suoraan yllä.){: .hands_on} # # Sra Run Selectorwearn earlier how to support our search results by using Entrez ’ s advanced syntax. Emme kuitenkaan hyödyntäneet sitä ylivoimaa, kun olimme Entrezissä. Sen sijaan käytimme yksinkertaista hakua ja sitten lähetimme kaikki tulokset Ajonvalitsimeen. Meillä ei ole vielä (lyhyttä) luetteloa tuloksista, joiden perusteella haluamme suorittaa analyysin. Mitä me teemme?*Käytämme Entreziä ja suorita-valitsinta, miten ne on suunniteltu käytettäväksi: * käytä Entrez-liitäntää rajataksesi tuloksesi kokoon, jonka Suorita-valitsin voi kuluttaa. * Lähetä nämä Entrez-tulokset Sra Run-valitsimeen * käytä Suorita-valitsimen paljon ystävällisempää käyttöliittymää arvoon 1. Helpommin ymmärtää tietoja meillä on 1. Kavenna tuloksia tuon tiedon avulla.> # # comment Run Selector is both more and less than Entrez>> Run Selector can do most, but not all of what Entrez search syntax can do. Suorita valitsin käyttää * faceted haku * tekniikka, joka on helppokäyttöinen ja tehokas, mutta jolla on luontainen rajat. Erityisesti Entrez toimii paremmin, kun etsitään ominaisuuksia, joilla on kymmeniä, satoja tai tuhansia erilaisia arvoja. Suorita valitsin toimii paremmin etsimällä attribuutteja alle 20 eri arvolla. Onneksi se kuvaa useimpia hakuja.{: .kommentti}Suorita – valitsinikkuna on jaettu useisiin paneeleihin:* **`Suodatinlista`**: vasemmassa yläkulmassa. Tässä tarkennamme hakua.* * * ’Select’**: yhteenveto siitä, mitä alun perin annettiin Selectorin ajamiseen, ja kuinka paljon siitä olemme valinneet tähän mennessä. (Ja toistaiseksi, emme ole valinneet mitään siitä.) Huomaa myös ärsyttävä, mutta silti harmaantunut, `Galaxy ’ nappi.* **` Found x Items ’ * * aluksi tämä on luettelo kohteista, jotka lähetetään suorittamaan valitsin Entrezistä. Tämä lista kutistuu, kun käytämme siihen suodattimia.!(../../ images / sra_run_valitsin.png)> # # kommentti miksi löytöjen määrä *nousi?* >> muista, että Entrez-rajapinta listaa SRA-kokeet (SRX-merkinnät). Suorita valintaluettelot *suoritukset* – sekvensointitiedot – ja on* yksi tai useampi * suorituksia per koe. Meillä on samat tiedot kuin ennenkin, nyt vain näemme sen tarkemmin.{: .kommentti}vasemmassa yläkulmassa oleva` suodattimien luettelo ’ näyttää sarakkeita tuloksissamme, joilla on joko jatkuvia numeerisia arvoja, tai 10 tai vähemmän (voit muuttaa tätä numeroa) erillisiä arvoja niissä. ** Selaa * * alas luettelon läpi valitse muutama suodatin. Kun suodatin on valittu, alla on * values* – ruutu, jossa luetellaan tämän suodattimen valinnat ja kunkin vaihtoehdon suorittamisten määrä. Nämä arvot / valinnat vedetään aineiston metadatasta. Kokeile * * valitsemalla * * muutama mielenkiintoinen kuulostava suodatin ja valitse** Valitse yksi tai useampi vaihtoehto kullekin suodattimelle. Kokeile * * valitsematta * * vaihtoehtoja ja suodattimia. Kun teet tämän, löydettyjen tulosten määrä vähenee tai kasvaa.> # # vinkkikärki: Käytä suotimia ymmärtääksesi paremmin tietoa>> suotimilla rajaat tarkasteltavat tiedot Galaxyyn lähetettäviksi, mutta ne ovat myös erinomainen tapa ymmärtää tietojasi:> ensin suodattimen valinta on helppo tapa nähdä sarakkeen arvoalue. Et ehkä pysty (https://www.google.com/search?q=sra+sirs_outcome), mutta voit mahdollisesti selvittää sen katsomalla, mitä arvoja siinä on.> toiseksi voit tutkia, miten eri sarakkeet liittyvät toisiinsa. Onko ”sirs_outcome” – arvojen ja ”disease_stage” – arvojen välillä yhteys?{: .vinkki}> # # hands_on Hands-on: rajaa tulokset käyttämällä Juoksuvalitsinta>> 1. Jos sinulla on Suodattimet päällä, **poista valinta** ne.> kun olet tehnyt tämän, ”suodattimien luettelon” alla ei ole yhtään *values* – ruutua.> 2. ** Kopioi ja liitä** tämä haku merkkijono ’Found Items’ hakukenttään.>> SRR11772204 tai SRR11597145 tai SRR11667145>> tämä käsi-poimitut ajot rajoittavat tuloksemme 3 ajoon eri maantieteellisestä jakaumasta.{: .hands_on}tämä vähentää ’Found Items’ lista kymmeniä tuhansia kulkee 3 kulkee (hallittavissa numero opetusohjelma!). Juoksuvalitsin ei ole vielä valmis. Huomaa, että ’Galaxy’ – painike on vielä harmaantunut. Olemme rajanneet vaihtoehtoja, mutta emme ole valinneet vielä mitään lähetettävää Galaxyyn.On mahdollista valita kaikki jäljellä olevat ajot * * klikkaamalla * * ensimmäisen sarakkeen yläreunassa olevaa valintamerkkiä. Voit poistaa kaiken valinnan** klikkaamalla * * X: ää.> # # hands_on Hands-on: Select runs and send to Galaxy>> 1. Valitse kaikki suoritukset** klikkaamalla * * X: ää.> Ja nyt ”Galaxy” – nappi on livenä.> 1. ** Klikkaa* * `Galaxy ’- painiketta ’Valitse’ – osiossa sivun yläreunassa.{: .hands_on}## Back in Galaxywen we click ’Galaxy’ In Run Selector several things happen. Ensinnäkin, se käynnistää uuden selaimen välilehti tai ikkuna, joka avautuu Galaxy. Näet * big green box * osoittaa, että kädenpuristus välillä SRA ja Galaxy oli onnistunut ja näet sitten uuden `SRA` työtä historiapaneelissa. Tämä ruutu voi alkaa harmaana / vireillä, mikä osoittaa, että siirto ei ole vielä alkanut, tai se voi mennä suoraan keltaiseen / käynnissä olevaan tai vihreään / tehtyyn.> # # hands_on Hands-on: Tarkastellaan uutta SRA-aineistoa>> 1. Kun` SRA ’ siirto on valmis, **klikkaa** datasetin galaxy-eye (eye) – kuvaketta.>> tämä näyttää aineiston Galaxyn keskipaneelissa.{: .hands_on}` SRA ’ – tietokokonaisuus ei ole sekvenssitietoa, vaan *metatietoa*, jota käytämme sekvenssitietojen saamiseksi SRA: lta. Tämä metadata peilaa tietoja näimme ajaa valitsimen ’Found Items’ osiossa. Metadata ei ole lopputietoa, jota haemme SRA: lta, mutta kaiken tuon metatiedon saaminen on usein hyödyllistä myöhemmissä analyysivaiheissa.Lets nyt käyttää että metadata hakea sekvenssitiedot Sra. SRA tarjoaa työkaluja kaikenlaisen tiedon, mukaan lukien sekvenssitiedot itse. Galaxy-työkalu ”Faster Download and Extract Reads in FASTQ”perustuu Sra (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump) – apuohjelmaan ja tekee juuri niin.– >
- etsi tarvittavat tiedot SRA: sta
- hands_on Hands-on: tehtävänkuvaus
- kommentti
- prosessi ja suodatin SraRunInfo.csv-tiedosto galaksissa
- hands_on Hands-on: Upload SraRunInfo.csv-tiedosto galaksiksi
- kommentti varo leikkauksia
- hands_on Hands-on: luodaan osajoukko tietoja
- tip Tip: Finding tools
- Download sequencing data with Faster Download and Extract Reads in FASTQ
- hands_on Hands-on: Task description
- mitä nyt?
- SARS-Cov-2-sekvensointitietojen Variaatioanalyysi
- kommentoi usegalaksia.* COVID-19-analyysiprojekti
- Get the reference genome data
- hands_on Hands-on: Get the reference genome data
- Tip: Tuominen linkkien kautta
- sovittimen trimmaus fastp: llä
- hands_on Hands-on: Task description
- linjaus kartan kanssa BWA-mem
- hands_on Hands-on: Align sequencing reads to reference genome
- Poista kaksoiskappaleet Markduplikaateilla
- hands_on Hands-on: Poista PCR-kaksoiskappaleet
- luo kohdistustilastot samtools-tilastoilla
- hands_on Hands-on: Luo tasaustilastot
- Realign lukee lofreq viterbi
- hands_on Hands-on: Realign reades around indels
- lisää indel-ominaisuuksia lofreq Insert indel-ominaisuuksilla
- hands_on hand-on: Lisää indel-ominaisuudet
- call variants käyttämällä lofreq-kutsuvariantteja
- hands_on Hands-on: Call variants
- Merkitkää variantin vaikutukset SnpEff eff: llä:
- hands_on Hands-on: Annotate variant effects
- Create table of variants using SnpSift Extract Fields
- hands_on Hands-on: Create table of variants
- summaa tiedot MultiQC: llä
- hands_on Hands-on: summary data
- johtopäätös
- keypoints Key points
- Frequently Asked Questions
- hyödyllinen kirjallisuus
- palaute
- vedoten tähän opetusohjelmaan
- details BibTeX
etsi tarvittavat tiedot SRA: sta
ensin täytyy löytää hyvä datajoukko jolla pelata. Sequence Read Archive (SRA) on Yhdysvaltain kansallisen terveysinstituutin (NIH) ylläpitämä unassembled readsin ensisijainen arkisto. SRA on hyvä paikka saada sekvensointitietoja, jotka ovat julkaisujen ja tutkimusten taustalla. Tehdään näin:
hands_on Hands-on: tehtävänkuvaus
- mene NCBI: n SRA-sivulle osoittamalla selaimesi https://www.ncbi.nlm.nih.gov/sra
- hakukenttään enter
SARS-CoV-2 Patient Sequencing From Partners / MGH(vaihtoehtoisesti klikkaamalla tätä linkkiä)
- verkkosivulla näkyy suuri määrä SRA-aineistoja (kirjoitushetkellä niitä oli 2 223). Tiedot ovat tutkimuksesta, jossa kuvailtiin SARS-CoV-2: n analyysia Bostonin alueella.
- lataa näitä tietokokonaisuuksia kuvaava metadata:
- klikkaamalla Lähetä: pudotusvalinta
- valitsemalla
File- muuttuva formaatti
RunInfo- napsauttamalla Luo tiedostoherestä on miltä sen pitäisi näyttää:
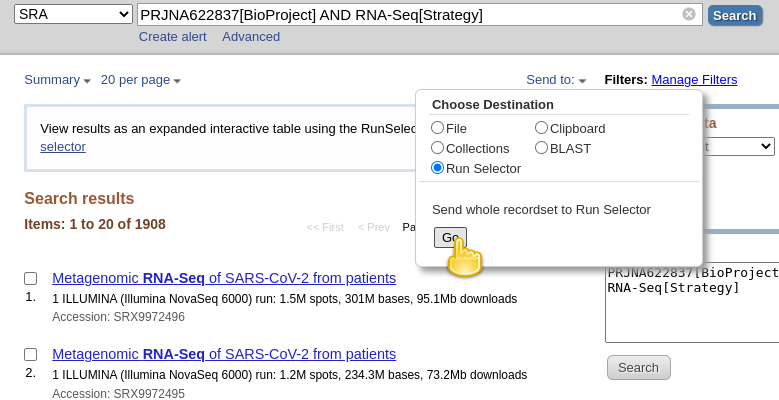
- tämä loisi melko suuren
SraRunInfo.csvtiedostonDownloadskansioon.
nyt kun olemme ladanneet tämän tiedoston, voimme mennä Galaxy-instanssiin ja aloittaa sen käsittelyn.
kommentti
huomaa, että juuri lataamamme tiedosto ei sekvensoi itse dataa. Pikemminkin se on metatietoa, joka kuvaa sekvensointilukujen ominaisuuksia. Suodatamme tämän luettelon vain muutamiin lisäyksiin, joita käytetään tämän opetusohjelman loppuosassa.
prosessi ja suodatin SraRunInfo.csv-tiedosto galaksissa
hands_on Hands-on: Upload SraRunInfo.csv-tiedosto galaksiksi
- Siirry Galaxy-instanssiin kuten johonkin usegalaxy.org, usegalaxy.eu, usegalaxy.org.au tai minkä tahansa muun. (Tämä opetusohjelma käyttää usegalaxy.org).
- klikkaa Upload Data-painiketta:
- ilmestyvässä valintaikkunassa klikkaa ”Valitse paikalliset tiedostot”-painiketta:

- Etsi ja valitse
SraRunInfo.csvtiedosto tietokoneeltasi- klikkaa Käynnistä-painiketta
- Sulje dialogi painamalla Sulje-painike
- voit nyt katsoa tiedoston sisältöä klikkaamalla Galaxy-eye (eye) – kuvaketta. Näet, että tämä tiedosto sisältää paljon tietoa yksittäisistä Sra-liittymistä. Tässä tutkimuksessa jokainen liittyminen vastaa yksittäistä potilasta, jonka näytteet sekvensoitiin.
Galaxy voi käsitellä kaikki yli 2 000 datajoukkoa, mutta jotta tämä opetusohjelma olisi siedettävä, on valittava pienempi osajoukko. Erityisesti aiemmat kokemuksemme tästä aineistosta osoittavat kaksi mielenkiintoista aineistoa SRR11954102 ja SRR12733957. Vedetään ne pois.
kommentti varo leikkauksia
alla olevassa käytännön osiossa käytetään leikkaustyökalua. Galaxyssa on historiallisista syistä kaksi leikkaustyökalua. Tässä esimerkissä käytetään työkalua, jonka koko nimi on leikatut sarakkeet taulukosta (cut). Sama logiikka pätee kuitenkin myös toiseen työkaluun. Se on yksinkertaisesti hieman erilainen käyttöliittymä.
hands_on Hands-on: luodaan osajoukko tietoja
- Etsi työkalu ”Valitse lauseketta vastaavat rivit työkalupaneelin suodatin-ja Lajitteluosassa.
tip Tip: Finding tools
Galaxy saattaa olla ylivoimainen määrä työkaluja asennettuna. Jos haluat löytää tietyn työkalun, Kirjoita työkalun nimi työkalupaneelin hakukenttään löytääksesi työkalun.
- varmista, että
SraRunInfo.csvjuuri lataamamme tieto on lueteltu työkalulomakkeen param-tiedoston ”Select lines from” – kentässä.- ”kuvion” kentässä merkitään seuraava lauseke →
SRR12733957|SRR11954102. Nämä ovat kaksi liittymistä, jotka halutaan erottaa toisistaan piipputunnuksella||tarkoittaaor: etsi rivit, jotka sisältävätSRR12733957taiSRR11954102.- klikkaa
Executenappia.- tämä luo tiedoston, joka sisältää kaksi riviä (no … yhtä riviä käytetään myös otsikkona, joten näyttää siltä, että tiedostossa on kolme riviä. Ei se mitään.)
- leikkaa ensimmäinen sarake tiedostosta työkalulla ”Cut”, jonka löydät työkaluruudun tekstinkäsittely-osiosta.
- varmista, että edellisen vaiheen tuottama aineisto on valittu työkalulomakkeen ”leikattava tiedosto” – kenttään.
- muuta ”rajattu”
Comma- ”kenttien luettelossa” valitse
Column: 1.- Hit
Executetämä tuottaa tekstitiedoston, jossa on vain kaksi riviä:
nyt kun meillä on tietoaineistojen tunnisteita me meidän on ladattava Sekvensointidata.
Download sequencing data with Faster Download and Extract Reads in FASTQ
hands_on Hands-on: Task description
- Faster Download and Extract Reads in FASTQ tool with the following parameters:
- ”select input type”:
List of SRA accession, one per line
- parametrin param-tiedosto” Sra-liittymisluettelo ”pitäisi osoittaa työkalun ulostulo” Cut ” edellisestä vaiheesta.
- klikkaa
Executenappia. Tämä ajaa työkalun, joka hakee sarjalukutiedot ajoille, jotka on lueteltuSRAdataset. Siihen voi mennä aikaa. Nyt voi olla hyvä hetki käydä kahvilla.- historiaraatiisi syntyy useita merkintöjä, kun lähetät tämän tehtävän:
Pair-end data (fasterq-dump): Sisältää Pariloppuisia tietokokonaisuuksia (jos saatavilla)Single-end data (fasterq-dump)Sisältää Yksipäisiä tietokokonaisuuksia (jos saatavilla)Other data (fasterq-dump)Sisältää parittomia tietokokonaisuuksia (jos saatavilla)fasterq-dump logsisältää tietoja työkalun toteutus
kolme ensimmäistä kohdetta ovat oikeastaan aineistokokoelmia. Galaxyn kokoelmat ovat loogisia aineiston ryhmittelyjä, jotka heijastavat kokeessa / analyysissä niiden välisiä semanttisia suhteita. Tässä tapauksessa työkalu luo erillisen kokoelman kullekin pariloppuiselle lukemiselle, yksittäiselle lukemiselle ja muille.Katso kokoelmat tutorials lisätietoja.
tutki kokoelmia klikkaamalla ensin kokoelman nimeä Historia-paneelissa. Tämä vie sinut kokoelman sisälle ja näyttää sen aineistot. Tämän jälkeen voit siirtyä takaisin historiasi ulkoiselle tasolle.
kun fasterq saa tiedonsiirron valmiiksi (kaikki ruudut ovat vihreitä / valmiita), olemme valmiita analysoimaan sen.
mitä nyt?
voit nyt analysoida haettua dataa millä tahansa sekvenssianalyysityökaluilla ja työnkuluilla Galaxyssa. SRA: lla on taustatiedot jokaisesta kuviteltavissa olevasta * – seq-kokeen tyypistä.
jos suoritit tämän opetusohjelman, mutta hait aineistoja, joista olit kiinnostunut, katso loput GTN-kirjastosta ideoita Galaxyn analysointiin.
Jos kuitenkin olet hakenut tämän opetusohjelman yllä olevissa esimerkeissä käytetyt aineistot, olet valmis suorittamaan alla olevan SARS-CoV-2-muunnosanalyysin.
SARS-Cov-2-sekvensointitietojen Variaatioanalyysi
opetusohjelman tässä osassa suoritamme varianttikutsun ja perusanalyysin yllä ladatuista aineistoista. Aloitamme lataamalla Wuhan-Hu-1 SARS-CoV-2-viitesekvenssin, sitten suoritamme sovittimen trimmauksen, linjauksen ja variantin kutsumisen ja lopuksi tarkastelemme joidenkin löydettyjen varianttien maantieteellistä jakautumista.
kommentoi usegalaksia.* COVID-19-analyysiprojekti
Tämä opetusohjelma käyttää osajoukkoa tiedoista ja kulkee covid19: n variaatioanalyysisektion läpi.galaksiprojekti.org.Tiedot covid19.galaxyproject.org sitä päivitetään jatkuvasti sitä mukaa kuin uusia tietokokonaisuuksia julkistetaan.
Get the reference genome data
the reference genome data for today is for SARS-CoV-2, ”Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome”, with the accession ID of NC_045512.2.
Tämä tieto on saatavissa Zenodosta seuraavasta linkistä.
hands_on Hands-on: Get the reference genome data
Import the following file into your history:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzTip: Tuominen linkkien kautta
- Kopioi linkin sijainti
- avaa Galaxy Upload Manager (galaxy-upload työkalupaneelin oikeassa yläkulmassa)
- Valitse Liitä/nouda data
- liitä linkki tekstikenttään
- paina Käynnistä
- sulje ikkunaehtoinen oletusarvo, Galaxy käyttää URL-osoitetta nimenä, joten nimeä tiedostot hyödyllisemmällä nimellä.
sovittimen trimmaus fastp: llä
sekvensointisovittimien poistaminen parantaa linjauksia ja variantikutsuja. fastp-työkalu tunnistaa automaattisesti laajalti käytetyt sekvensointisovittimet.
hands_on Hands-on: Task description
- fastp tool with the following parameters:
- ”Single-end or coupled reads”:
Paired Collection
- param-file ”Select coupled collection(s)”:
list_paired(Tuotos nopeammasta latauksesta ja otteesta lukee FASTQ-työkalussa)- ”Tulostusvaihtoehdoissa”:
- ”Tuotos JSON report”:
Yes
linjaus kartan kanssa BWA-mem
BWA-mem-työkalu on laajalti käytetty sekvenssialigner lyhyen lukusekvenssiin, kuten tässä opetusohjelmassa ANALYSOITAVIIN aineistoihin.
hands_on Hands-on: Align sequencing reads to reference genome
- Map with BWA-MEM tool with the following parameters:
- ”will you select a reference genome from your history or use a built-in index?”:
Use a genome from history and build index
- param-file ”käytä referenssisarjana seuraavaa aineistoa:
output(Syöttöaineisto)- ”yhden tai parin päässä lukee”:
Paired Collectionli> param-tiedosto”valitse paritettu kokoelma”:
output_paired_coll(fastp-työkalun tuotos)- ”aseta lukuryhmien tiedot?”:
Do not set- ”Select analysis mode”:
1.Simple Illumina mode
Poista kaksoiskappaleet Markduplikaateilla
Markduplikaatityökalu poistaa päällekkäiset sekvenssit, jotka ovat peräisin kirjaston valmistelu esineitä ja sekvensointi esineitä. On tärkeää poistaa nämä artefaktuaaliset sekvenssit yksittäisen molekyylin keinotekoisen yliedustuksen välttämiseksi.
hands_on Hands-on: Poista PCR-kaksoiskappaleet
- Markduplikoi työkalun seuraavilla parametreilla:
- param-tiedosto ”Select SAM/BAM dataset or dataset collection”:
bam_output(output of Map with BWA-MEM tool)- ”Jos tosi, älä kirjoita kaksoiskappaleita tulostustiedostoon sen sijaan, että kirjoittaisit ne asianmukaisilla lipuilla”:
Yes
luo kohdistustilastot samtools-tilastoilla
yllä olevan päällekkäisen merkintävaiheen jälkeen voimme luoda tilastotietoa LUOMASTAMME kohdistuksesta.
hands_on Hands-on: Luo tasaustilastot
- Samtools stats tool with the following parameters:
- param-file ”BAM file”:
outFile(output of Markduplikates tool)Set coverage distribution”:
NoOutput”:
One single summary file- ”filter by Sam flags”:
Do not filter- ”use a reference sequence”:
No- ”filter by regions”:
No
Realign lukee lofreq viterbi
Realign read tool korjaa lisäyksiä ja poistoja koskevia virheitä. Tämä on tarpeen, jotta voidaan tarkasti havaita variantteja.
hands_on Hands-on: Realign reades around indels
- Realign reades with lofreq tool with the following parameters:
- param-file ”Reades to realign”:
outFile(output of MarkDuplicates tool)- ”valitse vertailuperimän lähde”:
History
- param-file ”Reference”:
output(Input dataset)- In ”Advanced options”:
- ”miten käsitellä perusominaisuuksia 2?”:
Keep unchanged
lisää indel-ominaisuuksia lofreq Insert indel-ominaisuuksilla
Tämä vaihe lisää Indel-ominaisuuksia linjaustiedostoomme. Tämä on tarpeen, jotta voidaan kutsua variantteja käyttäen Lofreq-työkalulla varustettuja Kutsuvariantteja
hands_on hand-on: Lisää indel-ominaisuudet
- lisää indel-ominaisuudet lofreq-työkalulla seuraavin parametrein:
- param-file ”Reades”:
realigned(lähtö Readign reads tool)- ”Indel calculation approach”:
Dindel
- ”Valitse lähde referenssigenomille”:
History
- param-file ”reference”:
output(input dataset)
call variants käyttämällä lofreq-kutsuvariantteja
olemme nyt valmiita kutsumaan variantteja.
hands_on Hands-on: Call variants
- Call variants with lofreq tool with the following parameters:
- param-file ”Input reades in BAM format”:
output(output of Insert indel quality tool)- ”Valitse lähde reference genome”:
History
- param-file ”reference”:
output(input dataset)Call variants across”:
Whole referencetypes of variants to call”:
SNVs and indels- ”Variant calling parameters”:
Configure settings
- In ”Coverage”:
- ”Minimal coverage”:
50- In ”Base-calling”:
- ”vähimmäisperusteq”:
30vaihtoehtoisten perusteiden vähimmäisperusteq”:
30- in ”kartoituslaatu
20- ”variant Filter parameters”:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
tämän vaiheen ulostulo on kokoelma VCF-tiedostoja, jotka voidaan visualisoida genomiselaimella.
Merkitkää variantin vaikutukset SnpEff eff: llä:
nyt merkitään edellisessä vaiheessa kutsumamme variantit sillä vaikutuksella, joka niillä on SARS-CoV-2-genomiin.
hands_on Hands-on: Annotate variant effects
- SnpEff eff: tool with the following parameters:
- param-file ” Sequence changes (SNPs, MNPs, InDels)”:
variants(output of Call variants tool)- ”Output format”:
VCF (only if input is VCF)Create CSV report, usual for downstack analysis (-csvStats)”:
YesAnnotation options”: ` li> ”Filter output”: `
- ”filter out specific effects”:
No
tämän vaiheen ulostulo on vcf-tiedosto, johon on lisätty varianttiefektejä.
Create table of variants using SnpSift Extract Fields
we will now select different effects from the VCF and create a tabular file that is easily to understand for Human.
hands_on Hands-on: Create table of variants
- SnpSift Extract Fields tool with the following parameters:
- param-file ”Variant input file in VCF format”:
snpeff_output(output of SnpEff eff: tool)- ”fields to extract”:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- ”multiple field Separator”:
,- ”tyhjä kenttäteksti”:
.
voimme tarkastaa tulostetiedostot ja katsoa, onko tämän tiedoston variantteja kuvattu myös havainnoitavassa muistikirjassa, joka näyttää maantieteellisen SARS-cov-2-muunnossekvenssien
mielenkiintoisia variantteja ovat c-t-muunnos asemassa 14408 (14408c/t) srr11772204, 28144t/C srr11597145 ja 25563g/t srr11667145.
summaa tiedot MultiQC: llä
nyt tiivistämme analyysimme MultiQC: llä, joka tuottaa kauniin raportin datallemme.
hands_on Hands-on: summary data
- MultiQC tool with the following parameters:
- In ”Results”:
- param-toista ”Insert Results”
- ”Which tool was used generate lokeja?”:
fastp
- param-file”Output of fastp”:
report_json(fastp-työkalun tuotos)param-toista” Insert-tulokset ”
- ” mikä työkalu tuotti lokit?”:
Samtools
- In” Samtools output”:
- param-repeat” Insert Samtools output ”
- ” Type of Samtools output?”:
stats
- param-file ”Samtools stats output”:
output(tuotos Samtools stats tool)param-toista” Insert Results ”
- ” mikä työkalu tuotti lokit?”:
Picard
- In” Picard output”:
- param-repeat” Insert Picard output ”
- ” Type of Picard output?”:
Markdups- param-file ” Picard output:
metrics_file(Merkintätyökalun tuotos)param-toista” lisää tuloksia ”
- ” mikä työkalu tuotti lokit?”:
SnpEff
- Param-file ”Output of SnpEff”:
csvFile(output of SnpEff eff: tool)
johtopäätös
Onneksi olkoon, osaat nyt tuoda Sekvenssitietoja Sra: sta ja suorittaa esimerkkianalyysin näistä aineistoista.
keypoints Key points
Sra: n Sekvenssitiedot voidaan tuoda suoraan galaksiin
Frequently Asked Questions
Have questions about this tutorial? Tutustu Varianttianalyysin aiheeseen FAQ-sivulla nähdäksesi, onko kysymyksesi lueteltu siellä. Jos ei, kysy kysymyksesi GTN Gitter-Kanavalla tai Galaxy Help Forum
hyödyllinen kirjallisuus
lisätietoja, mukaan lukien linkit dokumentaatioon ja alkuperäisjulkaisuihin, tässä opetusohjelmassa kuvatuista työkaluista, analyysitekniikoista ja tulosten tulkinnasta löytyy täältä.
palaute
käytitkö tätä materiaalia kouluttajana? Voit vapaasti antaa meille palautetta siitä, miten se meni.
vedoten tähän opetusohjelmaan
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 alkaen NCBI: n Sequence Read Archive (Sra) to Galaxy: SARS-COV-2 variant analysis (Galaxy Training Materials). / training-material/topics/variant-analysis/tutorials/sars-cov-2 / tutorial.html Online; saatavilla tänään
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016 / J. cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}