Lo scopo di questo tutorial è quello di farvi conoscere l’elaborazione dei dati di sequenziamento di prossima generazione in Galaxy. Questo tutorial utilizza una chiamata variante COVID-19 da Illumina data, ma non si tratta di una chiamata variante di per sé.
Al termine di questo tutorial saprai:
- Come trovare i dati in SRA e trasferire queste informazioni a Galaxy
- Come eseguire l’elaborazione dei dati NGS di base in Galaxy, tra cui:
- Controllo di Qualità (QC) di Illumina dati
- Mappa
- la Rimozione di duplicati
- la Variante chiamata con
lofreq - Variante di annotazione
- Utilizzo di set di dati collezioni
- l’Importazione di dati a Jupyter
### Agenda>> In questo tutorial ci occuperemo di:>> 1. TOC > {: toc} > {: .agenda} # # Due percorsi attraverso questo tutorial Abbiamo creato duei traiettorie che puoi seguire attraverso questo tutorial.1. ** Traiettoria 1 * * – inizia con l’SRA di NCBI e cerca le adesioni disponibili → Inizia (#the-sequence-read-archive)2. ** Traiettoria 2 * * – bypass SRA di NCBI e iniziare con Galaxy direttamente. → Start (#back-in-galaxy)Si consiglia di iniziare con * * Traiettoria 2**.# La sequenza Read ArchiveThe (https://www.ncbi.nlm.nih.gov/sra) è l’archivio primario di *unassembled reads* per il (https://www.ncbi.nlm.nih.gov/). SRA è un ottimo posto per ottenere i dati di sequenziamento che sono alla base di pubblicazioni e studi.Questo tutorial copre come ottenere i dati di sequenza da SRA in Galaxy utilizzando una connessione diretta tra i due.> # # # comment Comment>> Si sente anche SRA denominato *Short Read Archive*, il suo nome originale.> {:.commento} # # Accesso SRASRA può essere raggiunto direttamente attraverso il suo sito web, o attraverso il pannello degli strumenti su Galaxy.> ### comment Comment>> Inizialmente l’opzione del pannello degli strumenti per accedere a SRA esiste solo sul (https://usegalaxy.org/). Il supporto per la connessione diretta a SRA sarà incluso nella versione 20.05 di Galaxy {:.commento}> ### hands_on Hands-on: Esplora SRA Entrez>> 1. Vai alla tua istanza Galaxy di scelta come uno dei (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) o qualsiasi altro. (Questo tutorial utilizza usegalaxy.org).> 1. Se la tua cronologia non è già vuota, inizia una nuova cronologia (vedi (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) per ulteriori informazioni sulle storie Galaxy)> 1. ** Fare clic su * * ‘Ottieni dati’ nella parte superiore del pannello degli strumenti.> 1. ** Fare clic su * * ‘SRA Server’ nell’elenco degli strumenti mostrato sotto `Get Data`.>Questo ti porta (https://www.ncbi.nlm.nih.gov/sra) can puoi anche iniziare direttamente dall’SRA. Nella parte superiore della pagina viene visualizzata una casella di ricerca. Prova a cercare qualcosa che ti interessa, come `dolphin` o `rene` o `dolphin rene` e poi **fare clic** il pulsante `Cerca`.>> Questo restituisce un elenco di esperimenti *SRA* che corrispondono alla stringa di ricerca. Gli esperimenti SRA, noti anche come voci *SRX*, contengono dati di sequenza da un particolare esperimento, nonché una spiegazione dell’esperimento stesso e di qualsiasi altro dato correlato. È possibile esplorare gli esperimenti restituiti cliccando sul loro nome. Vedi (https://www.ncbi.nlm.nih.gov/books/NBK56913/) nel (https://www.ncbi.nlm.nih.gov/books/n/helpsrakb/) per ulteriori informazioni.>>Quando si immette del testo nella casella di ricerca SRA, si sta utilizzando (https://www.ncbi.nlm.nih.gov/sra /docs/srasearch/). Entrez supporta sia ricerche di testo semplici, sia ricerche molto precise che controllano metadati specifici e utilizzano espressioni logiche arbitrariamente complesse. Entrez ti consente di scalare le tue ricerche da base a avanzate man mano che restringi le tue ricerche. La sintassi delle ricerche avanzate può sembrare scoraggiante, ma SRA fornisce una grafica (https://www.ncbi.nlm.nih.gov/sra /advanced/) per generare la sintassi specifica. E, come vedremo di seguito, il selettore di esecuzione SRA fornisce un’interfaccia utente ancora più amichevole per restringere i nostri dati selezionati.>> Gioca con l’interfaccia SRA Entrez, incluso Advanced query builder, per vedere se puoi identificare una serie di esperimenti SRA rilevanti per una delle tue aree di ricerca.{: .hands_on} > # # # hands_on Hands-on: Genera un elenco di esperimenti di corrispondenza usando Entrez>> Ora che hai una familiarità di base con SRA Entrez, troviamo le sequenze utilizzate in questo tutorial.>> 1. Se non sei già lì, * * naviga * * torna al (https://www.ncbi.nlm.nih.gov/sra> 1. ** Cancella * * qualsiasi testo di ricerca dalla casella di ricerca.> 1. ** Digitare* * `sars-cov-2 ‘nella casella di ricerca e * * fare clic* * ‘Cerca’.> Questo restituisce una lunga lista di esperimenti SRA che corrispondono alla nostra ricerca, e quella lista è troppo lunga per essere utilizzata in un esercizio di esercitazione. A questo punto potremmo usare il costruttore di query Entrez avanzato che abbiamo appreso sopra.> Ma non lo faremo. Invece consente di inviare il *troppo lungo per un tutorial* lista risultati che abbiamo al selettore di esecuzione SRA, e utilizzare la sua interfaccia più amichevole per restringere i nostri risultati.>>!(../../ images / sra_entrez.png){: .hands_on} > # # # hands_on Hands-on: Passare da Entrez a SRA Run Selector>> Visualizza i risultati come una tabella interattiva espansa utilizzando RunSelector.>> 1. Fare clic su Invia risultati per eseguire selettore, che appare in una casella nella parte superiore dei risultati della ricerca.>>!(../../ images / sra_entrez_result.png) >>> # # # suggerimento Cosa succede se non vedi il link Run Selector?>>>> Potresti aver notato questo testo in precedenza mentre stavi esplorando la ricerca di Entrez. Questo testo appare solo una parte del tempo, quando il numero di risultati di ricerca rientra in una finestra abbastanza ampia. Non lo vedrai se hai solo pochi risultati e non lo vedrai se hai più risultati di quelli che il selettore di esecuzione può accettare.>>>> *È necessario arrivare a eseguire Selettore per inviare i risultati a Galaxy.* Cosa succede se non si dispone di risultati sufficienti per attivare questo collegamento viene mostrato? In tal caso si chiama get al selettore Esegui **cliccando** sul menu a tendina `Invia a` in alto a destra del pannello dei risultati. Per arrivare a Eseguire Selettore, * * selezionare * * ‘Esegui selettore’ e poi * * fare clic* * il pulsante ‘Go’.>!(../../ images / sra_entrez_send_to.png) > {: .tip} >>> 1. ** Fare clic su * * ‘Invia risultati per eseguire selettore’ nella parte superiore del pannello dei risultati di ricerca. (Se non vedi questo link, vedi il commento direttamente sopra.){: .hands_on} # # SRA Run selectorabbiamo imparato in precedenza come restringere i risultati della ricerca utilizzando la sintassi avanzata di Entrez. Tuttavia, non abbiamo approfittato di quel potere quando eravamo a Entrez. Invece abbiamo usato una semplice ricerca e poi inviato tutti i risultati al selettore Run. Non abbiamo ancora l’elenco (breve) dei risultati su cui vogliamo eseguire l’analisi. *Cosa stiamo facendo?* Stiamo usando Entrez e il Selettore Run come sono progettati per essere utilizzati: * Utilizzare l’interfaccia Entrez per restringere i risultati fino a una dimensione che il selettore Run può consumare. * Inviare questi risultati Entrez al selettore di esecuzione SRA * Utilizzare l’interfaccia molto più amichevole del selettore di esecuzione a 1. Più facilmente capire i dati che abbiamo 1. Restringere quei risultati usando quella conoscenza.> # # # comment Run Selector è sia più che meno di Entrez>> Run Selector può fare la maggior parte, ma non tutto ciò che Entrez search syntax può fare. Run selector utilizza la tecnologia * ricerca sfaccettata * che è facile da usare e potente, ma che ha limiti intrinseci. In particolare, Entrez funzionerà meglio durante la ricerca su attributi che hanno decine, centinaia o migliaia di valori diversi. Run Selector funzionerà meglio cercando attributi con meno di 20 valori diversi. Fortunatamente, questo descrive la maggior parte delle ricerche.{: .commento}La finestra Run Selector è divisa in diversi pannelli: * * * ‘Lista filtri’**: Nell’angolo in alto a sinistra. Questo è dove affinare la nostra ricerca.* * * ` Select’**: Un riassunto di ciò che è stato inizialmente passato a Run Selector, e quanto di quello che abbiamo selezionato finora. (E finora, non abbiamo selezionato nulla di tutto ciò.) Si noti anche l’allettante, ma ancora in grigio, pulsante `Galaxy’.** * ` Trovato x elementi ‘ * * Inizialmente, questo è l’elenco degli elementi inviati per eseguire Selettore da Entrez. Questo elenco si ridurrà man mano che applichiamo i filtri ad esso.!(../../ images / sra_run_selector.png) > # # # commento Perché il numero di elementi trovati * è aumentato?* >> Ricordiamo che l’interfaccia Entrez elenca gli esperimenti SRA (voci SRX). Run Selector lists * runs * – set di dati di sequenziamento – e ci sono* uno o più * runs per esperimento. Abbiamo gli stessi dati di prima, ora li vediamo solo nei minimi dettagli.{: .commento}La ‘Lista filtri’ in alto a sinistra mostra colonne nei nostri risultati che hanno valori numerici continui, o 10 o meno (è possibile modificare questo numero) valori distinti in essi. ** Scorrere * * verso il basso attraverso l’elenco selezionare alcuni dei filtri. Quando viene selezionato un filtro, viene visualizzata una casella* valori*, che elenca le opzioni per questo filtro e il numero di esecuzioni con ciascuna opzione. Questi valori / opzioni vengono estratti dai metadati del set di dati. Prova * * selezione * * alcuni filtri dal suono interessanti e poi** selezionare * * una o più opzioni per ogni filtro. Prova** deselezionare * * opzioni e filtri. Mentre lo fai, il numero di risultati trovati diminuirà o aumenterà.> # # # tip Tip: Utilizza i Filtri per comprendere meglio i dati>> Filtri sono come potete restringere il set di dati presi in considerazione per l’invio di Galaxy, ma sono anche un ottimo modo per comprendere i tuoi dati:> in Primo luogo, la selezione di un filtro è un modo semplice per vedere la gamma di valori in una colonna. Potresti non essere in grado di (https://www.google.com/search?q=sra+sirs_outcome), ma puoi eventualmente capirlo vedendo quali valori ci sono in esso.> In secondo luogo, puoi esplorare come le diverse colonne si relazionano tra loro. Esiste una relazione tra i valori “sirs_outcome” e i valori “disease_stage”?{: .tip} > # # # hands_on Hands-on: Restringere i risultati utilizzando Run Selector>> 1. Se hai dei filtri attivati, * * deselezionali**.> Una volta fatto questo, non ci saranno caselle *values* che appaiono sotto la `Lista filtri`.> 2. ** Copia e incolla * * questa stringa di ricerca nella casella di ricerca “Elementi trovati”.>> SRR11772204 O SRR11597145 O SRR11667145>> Questo, raccolte a mano set di esecuzioni limiti i nostri risultati a 3 piste da diversa distribuzione geografica.{: .hands_on} Questo riduce la tua lista di “Elementi trovati” da decine di migliaia di esecuzioni a 3 esecuzioni (un numero gestibile per un tutorial!). Ma non abbiamo ancora finito con Run Selector. Si noti che il pulsante` Galaxy ‘ è ancora in grigio. Abbiamo ristretto le nostre opzioni, ma non abbiamo ancora selezionato nulla da inviare a Galaxy.E ‘ possibile selezionare ogni corsa rimanente **cliccando** il segno di spunta nella parte superiore della prima colonna. È possibile deselezionare tutto * * cliccando * * la ‘X’.> ### hands_on Hands-on: selezionare le corse e inviare a Galaxy>> 1. Selezionare tutte le corse * * cliccando * * la ‘X’.> E ora, il pulsante `Galaxy` è attivo.> 1. ** Fare clic* * il pulsante ‘Galaxy’ nella sezione ‘Seleziona’ nella parte superiore della pagina.{: .hands_on} # # Torna in Galaxyquando clicchiamo ‘Galaxy’ nel Selettore Run accadono diverse cose. Innanzitutto, lancia una nuova scheda o finestra del browser che si apre in Galaxy. Vedrai la * grande scatola verde * che indica che la stretta di mano tra SRA e Galaxy ha avuto successo e vedrai un nuovo lavoro `SRA` nel tuo pannello di cronologia. Questa casella può iniziare come grigio / in sospeso, indicando che il trasferimento non è ancora iniziato, o può andare direttamente in giallo / in esecuzione o in verde / fatto.> # # # hands_on Hands-on: Esaminare il nuovo set di dati SRA>> 1. Una volta completato il trasferimento` SRA’, **fare clic** sull’icona galaxy-eye (eye) del set di dati.>> Visualizza il set di dati nel pannello centrale di Galaxy.{: .hands_on}Il set di dati ‘ SRA ‘ non è dati di sequenza, ma piuttosto *metadati* che useremo per ottenere i dati di sequenza da SRA. Questi metadati rispecchiano le informazioni che abbiamo visto nella sezione “Elementi trovati” del Selettore di esecuzione. I metadati non sono i dati finali che stiamo cercando da SRA, ma avere tutti quei metadati è spesso utile nelle fasi di analisi successive.Consente ora di utilizzare i metadati per recuperare i dati di sequenza da SRA. SRA fornisce strumenti per l’estrazione di tutti i tipi di informazioni, compresi i dati di sequenza stessa. Lo strumento Galaxy ‘Faster Download and Extract Reads in FASTQ’ si basa sull’utilità SRA (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump) e fa proprio questo.– >
- Trova i dati necessari in SRA
- hands_on Hands-on: Descrizione dell’attività
- commento Commento
- Elabora e filtra SraRunInfo.file csv in Galaxy
- hands_on Hands-on: Carica SraRunInfo.file csv in Galaxy
- commento Attenzione ai tagli
- hands_on Hands-on: Creazione di un sottoinsieme di dati
- suggerimento Suggerimento: Trovare strumenti
- Scarica i dati di sequenziamento con download ed estratto più veloci Legge in FASTQ
- hands_on Hands-on: Descrizione dell’attività
- E ora?
- Analisi della variazione dei dati di sequenziamento SARS-Cov-2
- commenta l’usegalaxy.* COVID-19 analysis project
- Ottieni i dati del genoma di riferimento
- hands_on Hands-on: Ottieni i dati del genoma di riferimento
- Suggerimento: Importazione tramite link
- Ritaglio adattatore con fastp
- hands_on Hands-on: Descrizione dell’attività
- Allineamento con la Mappa BWA-MEM
- hands_on Hands-on: Align sequencing legge al genoma di riferimento
- Rimuovi i duplicati con MarkDuplicates
- hands_on Hands-on: Rimuovere i duplicati PCR
- Generare allineamento con le statistiche di Samtools statistiche
- hands_on Hands-on: Generare allineamento delle statistiche
- Riallineare legge con lofreq viterbi
- hands_on Hands-on: Riallineare legge intorno indels
- Aggiungi qualità indel con lofreq Inserisci qualità indel
- hands_on Hands-on: Aggiungere indel qualità
- Chiamate Varianti utilizzando lofreq Chiamata varianti
- hands_on Hands-on: Chiamata varianti
- Annota gli effetti delle varianti con SnpEff eff:
- hands_on Hands-on: Annota effetti varianti
- Crea una tabella di varianti usando SnpSift Extract Fields
- hands_on Hands-on: Creare la tabella di varianti
- Riassumere i dati con MultiQC
- hands_on Hands-on: Riepiloga i dati
- Conclusione
- punti chiave Punti chiave
- Domande frequenti
- Letteratura utile
- Feedback
- Citando questo Tutorial
- details BibTeX
Trova i dati necessari in SRA
Per prima cosa dobbiamo trovare un buon set di dati con cui giocare. Il Sequence Read Archive (SRA) è l’archivio principale di letture non assemblate gestito dal National Institutes of Health (NIH) degli Stati Uniti. SRA è un ottimo posto per ottenere i dati di sequenziamento che sono alla base di pubblicazioni e studi. Facciamolo:
hands_on Hands-on: Descrizione dell’attività
- Vai alla pagina SRA di NCBI puntando il browser ahttps://www.ncbi.nlm.nih.gov/sra
- Nella casella di ricerca inserisci
SARS-CoV-2 Patient Sequencing From Partners / MGH(In alternativa, è sufficiente fare clic su questo link)
- La pagina web mostrerà un gran numero di set di dati SRA (al momento della scrittura c’erano 2.223). Si tratta di dati provenienti da uno studio che descrive l’analisi di SARS-CoV-2 nella zona di Boston.
- Scarica i metadati che descrivono questi set di dati da:
- facendo clic su Invia a: menu a discesa
- Selezionando
File- Cambiando il formato a
RunInfo- Facendo clic su Crea filequi è come dovrebbe apparire:
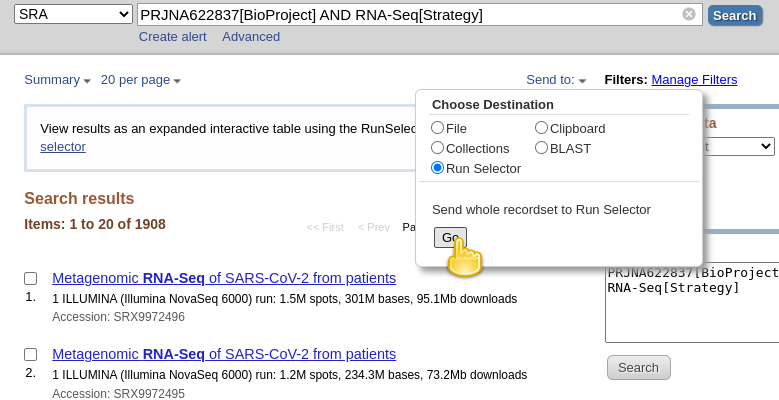
- Questo creerebbe un file piuttosto grande
SraRunInfo.csvnella cartellaDownloads.
Ora che abbiamo scaricato questo file possiamo andare su un’istanza Galaxy e iniziare a elaborarlo.
commento Commento
Si noti che il file che abbiamo appena scaricato non è il sequenziamento dei dati stessi. Piuttosto, si tratta di metadati che descrivono le proprietà di sequenziamento legge. Filtreremo questo elenco fino a poche adesioni che verranno utilizzate nel resto di questo tutorial.
Elabora e filtra SraRunInfo.file csv in Galaxy
hands_on Hands-on: Carica SraRunInfo.file csv in Galaxy
- Vai alla tua istanza Galaxy di scelta come uno dei usegalaxy.org, usegalaxy.eu, usegalaxy.org.au o qualsiasi altro. (Questo tutorial utilizza usegalaxy.org).
- Fare clic sul pulsante Carica dati:
- Nella finestra di dialogo che appare fare clic su “Scegli file locali” pulsante:

- Trovare e selezionare
SraRunInfo.csvfile dal tuo computer- fare Clic sul pulsante Start
- Chiudere la finestra di dialogo premendo il pulsante di chiusura
- È possibile visualizzare il contenuto di questo file cliccando galaxy-eye (occhio) icona. Vedrai che questo file contiene molte informazioni sulle singole adesioni SRA. In questo studio ogni adesione corrisponde a un singolo paziente i cui campioni sono stati sequenziati.
Galaxy può elaborare tutti i 2.000 + set di dati, ma per rendere questo tutorial sopportabile abbiamo bisogno di selezionato un sottoinsieme più piccolo. In particolare la nostra precedente esperienza con questi dati mostra due interessanti dataset SRR11954102 e SRR12733957. Quindi, tiriamoli fuori.
commento Attenzione ai tagli
La sezione pratica qui sotto utilizza lo strumento di taglio. Ci sono due strumenti di taglio in Galaxy a causa di ragioni storiche. Questo esempio utilizza lo strumento con il nome completo Taglia colonne da una tabella (taglia). Tuttavia, la stessa logica si applica all’altro strumento. Ha semplicemente un’interfaccia leggermente diversa.
hands_on Hands-on: Creazione di un sottoinsieme di dati
- Trova strumento “Seleziona linee che corrispondono a un’espressione” strumento nella sezione Filtra e Ordina del pannello strumenti.
suggerimento Suggerimento: Trovare strumenti
Galaxy potrebbe avere una quantità enorme di strumenti installati. Per trovare uno strumento specifico digitare il nome dello strumento nella casella di ricerca del pannello strumenti per trovare lo strumento.
- Assicurati che il set di dati
SraRunInfo.csvche abbiamo appena caricato sia elencato nel campo “Seleziona linee da” del modulo strumento.- Nel campo “il modello” immettere la seguente espressione →
SRR12733957|SRR11954102. Queste sono due accessioni che vogliamo trovare separate dal simbolo pipe|. Il|significaor: trova linee contenentiSRR12733957oSRR11954102.- Fare clic su
Executepulsante.- Questo genererà un file contenente due righe (beh one una riga è anche usata come intestazione, quindi apparirà che il file ha tre righe. Va bene.)
- Taglia la prima colonna dal file usando lo strumento” Taglia”, che troverai nella sezione Manipolazione del testo del riquadro degli strumenti.
- Assicurarsi che il set di dati prodotto dal passaggio precedente sia selezionato nel campo” File da tagliare ” del modulo strumento.
- Cambia “Delimitato da” in
Comma- In “Elenco di campi” seleziona
Column: 1.- Premi
ExecuteQuesto produrrà un file di testo con solo due righe:SRR12733957SRR11954102
Ora che abbiamo gli identificatori di set di dati si vuole, è necessario scaricare l’effettiva dati di sequenziamento.
Scarica i dati di sequenziamento con download ed estratto più veloci Legge in FASTQ
hands_on Hands-on: Descrizione dell’attività
- Download ed estratto più veloci legge nello strumento FASTQ con i seguenti parametri:
- “select input type”:
List of SRA accession, one per line
- Il parametro param-file” sra accession list “dovrebbe puntare l’output dello strumento” Cut” dal passaggio precedente.
- Fare clic sul pulsante
Execute. Verrà eseguito lo strumento, che recupera i set di dati di lettura della sequenza per le esecuzioni elencate nel set di datiSRA. Potrebbe volerci del tempo. Quindi questo potrebbe essere un buon momento per prendere il caffè.- Nel pannello cronologia vengono create diverse voci quando si invia questo lavoro:
Pair-end data (fasterq-dump): Contiene Paired-end di set di dati (se disponibile)Single-end data (fasterq-dump)Contiene Singolo-end di set di dati (se disponibile)Other data (fasterq-dump)Contiene Spaiato di set di dati (se disponibile)fasterq-dump logContiene Informazioni sull’esecuzione dei tool
I primi tre elementi sono effettivamente collezioni di set di dati. Le raccolte in Galaxy sono raggruppamenti logici di set di dati che riflettono le relazioni semantiche tra loro nell’esperimento / analisi. In questo caso lo strumento crea una raccolta separata ciascuna per letture accoppiate, letture singole e altro.Vedere le collezioni tutorial per più.
Esplora le raccolte facendo prima clic sul nome della raccolta nel pannello cronologia. Questo ti porta all’interno della raccolta e ti mostra i set di dati in esso contenuti. È quindi possibile tornare al livello esterno della cronologia.
Una volta che fasterq termina il trasferimento dei dati (tutte le caselle sono verdi / fatte), siamo pronti per analizzarlo.
E ora?
È ora possibile analizzare i dati recuperati utilizzando tutti gli strumenti di analisi di sequenza e flussi di lavoro in Galaxy. SRA detiene i dati di supporto per ogni tipo immaginabile di * – seq esperimento.
Se hai eseguito questo tutorial, ma hai recuperato i set di dati che ti interessavano, vedi il resto della libreria GTN per idee su come analizzare in Galaxy.
Tuttavia, se hai recuperato i set di dati utilizzati negli esempi di questo tutorial sopra, allora sei pronto per eseguire l’analisi della variante SARS-CoV-2 di seguito.
Analisi della variazione dei dati di sequenziamento SARS-Cov-2
In questa parte del tutorial eseguiremo la chiamata variante e l’analisi di base dei set di dati scaricati sopra. Inizieremo scaricando la sequenza di riferimento Wuhan-Hu-1 SARS-CoV-2, quindi eseguiremo il trimming dell’adattatore, l’allineamento e la chiamata della variante e infine esamineremo la distribuzione geografica di alcune delle varianti trovate.
commenta l’usegalaxy.* COVID-19 analysis project
Questo tutorial utilizza un sottoinsieme dei dati e passa attraverso l’analisi della variazionesezione di covid19.galaxyproject.org.I dati per covid19.galaxyproject.org isbeing aggiornato continuamente come nuovi set di dati sono resi pubblici.
Ottieni i dati del genoma di riferimento
I dati del genoma di riferimento per oggi sono per SARS-CoV-2, “Grave sindrome respiratoria acuta coronavirus 2 isolato Wuhan-Hu-1, genoma completo”, con l’ID di adesione di NC_045512.2.
Questi dati sono disponibili da Zenodo utilizzando il seguente link.
hands_on Hands-on: Ottieni i dati del genoma di riferimento
Importa il seguente file nella cronologia:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzSuggerimento: Importazione tramite link
- Copia il percorso del link
- Aprire il Galaxy Upload Manager (galaxy-di caricamento in alto a destra del pannello degli strumenti)
- Selezionare Incolla/Recupero Dati
- Incolla il link nel campo di testo
- Premere Start
- Chiudere la windowBy di default, Galaxy utilizza l’URL come il nome, quindi rinominare il file con un nome più utile.
Ritaglio adattatore con fastp
La rimozione degli adattatori di sequenziamento migliora gli allineamenti e la chiamata variante. lo strumento fastp può rilevare automaticamente gli adattatori di sequenziamento ampiamente utilizzati.
hands_on Hands-on: Descrizione dell’attività
- strumento fastp con i seguenti parametri:
- “Single-end or paired reads”:
Paired Collection
- param-file”Select paired collection(s)”:
list_paired(uscita di rendere più Veloce il Download e l’Estratto di Legge in FASTQ strumento)- In “Opzioni di Output”:
- “Output JSON report”:
Yes
Allineamento con la Mappa BWA-MEM
BWA-MEM strumento è ampiamente utilizzato sequenza allineatore per breve lettura di sequenziamento di set di dati come quelli che stiamo analizzando in questo tutorial.
hands_on Hands-on: Align sequencing legge al genoma di riferimento
- Mappa con strumento BWA-MEM con i seguenti parametri:
- ” Vuoi selezionare un genoma di riferimento dalla vostra storia o utilizzare un indice integrato?”:
Use a genome from history and build index
- param-file “Utilizza il seguente set di dati come la sequenza di riferimento”:
output(set di dati di Input)- “Singoli o Associati-end legge”:
Paired Collection
- param-file “, Selezionare una coppia di raccolta”:
output_paired_coll(uscita di fastp strumento)- “Set di leggere le informazioni sui gruppi?”:
Do not set- “Select analysis mode”:
1.Simple Illumina mode
Rimuovi i duplicati con MarkDuplicates
Lo strumento MarkDuplicates rimuove le sequenze duplicate originarie dalla preparazione della libreria artefatti e artefatti di sequenziamento. È importante rimuovere queste sequenze artefatte per evitare una sovrarappresentazione artificiale di una singola molecola.
hands_on Hands-on: Rimuovere i duplicati PCR
- Strumento MarkDuplicates con i seguenti parametri:
- param-file “, Selezionare SAM/BAM dataset o un set di dati di raccolta”:
bam_output(uscita di Mappa con BWA-MEM strumento)- Se vero non scrivere duplicati per il file di output, invece di scrivere a loro con opportuni flag set”:
Yes
Generare allineamento con le statistiche di Samtools statistiche
Dopo il duplicato del contrassegno passo sopra, siamo in grado di generare statistiche circa l’allineamento abbiamo generato.
hands_on Hands-on: Generare allineamento delle statistiche
- Samtools statistiche strumento con i seguenti parametri:
- param-file di “BAM” file”:
outFile(uscita di MarkDuplicates strumento)- “Set di copertura distribuzione”:
No- “Uscita”:
One single summary file- “Filter by SAM il flag”:
Do not filter- Usare una sequenza di riferimento”:
No- Filtro “da parte delle regioni”:
No
Riallineare legge con lofreq viterbi
Riallineare legge strumento corregge disallineamenti intorno inserimenti e cancellazioni. Questo è necessario per rilevare con precisione le varianti.
hands_on Hands-on: Riallineare legge intorno indels
- Riallineare legge con lofreq strumento con i seguenti parametri:
- param-file “, si Legge a riallineare”:
outFile(uscita di MarkDuplicates strumento)- “selezionare la sorgente per il riferimento del genoma”:
History
- param-file”Reference”:
output(Set di dati di input)- In “Opzioni avanzate”:
- ” Come gestire le qualità di base di 2?” :
Keep unchanged
Aggiungi qualità indel con lofreq Inserisci qualità indel
Questo passaggio aggiunge qualità indel nel nostro file di allineamento. Questo è necessario per chiamare varianti usando le varianti di chiamata con lo strumento lofreq
hands_on Hands-on: Aggiungere indel qualità
- Inserire indel qualità con lofreq strumento con i seguenti parametri:
- param-file di “Legge”:
realigned(uscita di Riallineare legge tool)- “Indel metodo di calcolo”:
Dindel
- “selezionare la sorgente per il genoma di riferimento”:
History
- param-file di “Riferimento”:
output(set di dati di Input)
Chiamate Varianti utilizzando lofreq Chiamata varianti
ora Siamo pronti per chiamare varianti.
hands_on Hands-on: Chiamata varianti
- Chiamate varianti con lofreq strumento con i seguenti parametri:
- param-file “Input legge in BAM” formato:
output(uscita di Inserire indel qualità di strumento)- “selezionare la sorgente per il genoma di riferimento”:
History
- param-file di “Riferimento”:
output(set di dati di Input)- “Chiamata varianti tra i”:
Whole reference- “Tipi di varianti a chiamata”:
SNVs and indels- “Variant calling parameters”:
Configure settings
- In “Coverage”:
- “Minimal coverage”:
50- In “Base-calling quality”:
- “Minimum baseQ”:
30- “Minimum baseQ for alternate bases”:
30- In “Mapping quality”:
- “Minimum mapping quality”:
20- “Variant filter parameters”:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
L’output di questo passaggio è una raccolta di file VCF che possono essere visualizzati in un browser genoma.
Annota gli effetti delle varianti con SnpEff eff:
Ora annoteremo le varianti che abbiamo chiamato nel passaggio precedente con l’effetto che hanno sul genoma SARS-CoV-2.
hands_on Hands-on: Annota effetti varianti
- SnpEff eff: strumento con i seguenti parametri:
- param-file ” Sequence changes (SNPs, MNPs, InDels)”:
variants(uscita di Call varianti strumento)- “formato di Uscita”:
VCF (only if input is VCF)- “Creare report CSV, utile per l’analisi a valle (-csvStats)”:
Yes- “Annotazione” opzioni: `
- “l’uscita del Filtro”: `
- “Filtrare Effetti specifici”:
No
L’output di questa fase è un file VCF con l’aggiunta di variante effetti.
Crea una tabella di varianti usando SnpSift Extract Fields
Ora selezioneremo vari effetti dal VCF e creeremo un file tabulare che è più facile da capire per gli esseri umani.
hands_on Hands-on: Creare la tabella di varianti
- SnpSift Estrarre i Campi strumento con i seguenti parametri:
- param-file di “Variante file di input in formato VCF”:
snpeff_output(uscita di SnpEff fep: strumento)- “Campi da estrarre”:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- “più separatore di campo”:
,- “vuoto campo di testo”:
.
Possiamo controllare i file di output e vedere controllare se le Varianti di questo file sono descritti anche in un osservabili notebook che mostra la distribuzione geografica del SARS-CoV-2 variante di sequenze
Interessante varianti includono i C a T variante alla posizione 14408 (14408C/T) in SRR11772204, 28144T/C in SRR11597145 e 25563G/T in SRR11667145.
Riassumere i dati con MultiQC
Ora riassumiamo la nostra analisi con MultiQC, che genera un bel rapporto per i nostri dati.
hands_on Hands-on: Riepiloga i dati
- Strumento MultiQC con i seguenti parametri:
- In “Risultati”:
- param-repeat “Inserisci risultati”
- “Quale strumento è stato utilizzato genera log?” :
fastp
- param-file ” Uscita di fastp”:
report_json(output dello strumento fastp)- param-repeat” Inserisci risultati “
- ” Quale strumento è stato utilizzato genera log?” :
Samtools
- In” Uscita Samtools”:
- param-repeat” Inserisci uscita Samtools “
- ” Tipo di uscita Samtools?” :
stats
- param-file ” Samtools stats output”:
output(output dello strumento statistiche Samtools)- param-repeat “Inserisci risultati”
- “Quale strumento è stato utilizzato genera log?” :
Picard
- In” Uscita Picard”:
- param-repeat” Inserisci uscita Picard “
- ” Tipo di uscita Picard?”:
Markdups- param-file ” Uscita Picard”:
metrics_file(output dello strumento MarkDuplicates)- param-repeat “Inserisci risultati”
- “Quale strumento è stato utilizzato genera log?”:
SnpEff
- param-file di Output “di SnpEff”:
csvFile(uscita di SnpEff fep: strumento)
Conclusione
Congratulazioni, adesso sai come sequenza di importazione dati da SRA e come eseguire un esempio di analisi su questi dati.
punti chiave Punti chiave
I dati di sequenza nell’SRA possono essere importati direttamente in Galaxy
Domande frequenti
Hai domande su questo tutorial? Controlla la pagina delle FAQ per l’argomento di analisi delle varianti per vedere se la tua domanda è elencata lì. In caso contrario, fai la tua domanda sul canale Gitter GTN o sul Forum di aiuto Galaxy
Letteratura utile
Ulteriori informazioni, inclusi i link alla documentazione e alle pubblicazioni originali, riguardanti gli strumenti, le tecniche di analisi e l’interpretazione dei risultati descritti in questo tutorial possono essere trovate qui.
Feedback
Hai usato questo materiale come istruttore? Sentitevi liberi di darci un feedback su come è andata.
Citando questo Tutorial
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 Da Sequence Read Archive (SRA) di NCBI a Galaxy: SARS-COV-2 variant analysis (Galaxy Training Materials). / formazione-materiale / argomenti / variante-analisi / tutorial / sars-cov-2 / tutorial.html online; accessibile OGGI
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016 / j. cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}