Aminoacidi, nucleotidi o qualsiasi altro carattere evolutivo sono sostituiti daaltri ad un certo ritmo. Per esempio, immaginate una catena evolutiva con threepossible stati, A, B e C. Se la sostituzione del modello è reversibile, therewill tre transizione tassi, di Un<>B, B<>C e Un<>C.
Supponiamo che i tassi siano 1, 1 e 0 rispettivamente in unità di sostituzione per 100 caratteri per unità di tempo. Dopo un’unità di tempo, in una sequenza lunga 300 caratteri originariamente composta ugualmente da As, Bs e Cs, ci aspettiamo che ci sia stata una sostituzione da A a B e una sostituzione da B a C. Se stiamo confrontando due sequenze omologhe negli organismi viventi, perché un’unità di tempo è passata per entrambe le sequenze, ci aspetteremmo due A a B e due B a Csubstitutions tra le sequenze attuali.
Non importa per quanto tempo eseguiamo questo processo, non ci sarà mai una sostituzione diretta di A da C. Non ci sarà mai una sostituzione da A a C sotto il cosiddetto modello di siti infiniti, dove non può verificarsi più di una sostituzione in un singolo sito.
Tuttavia, poiché le sostituzioni da A a B e da B a C sono comuni, sotto un modello di sitesmodel finito alla fine B sarà sostituito da C in un sito in cui A è stato precedentemente sostituito da B. Questa sostituzione indiretta di A con C (o equivalentemente in un modello reversibile, C con A) diventa più probabile più lungo è il periodo di
Ho simulato l’evoluzione della sequenza in base allo scenario precedente, eseguendo la simulazione per 10 unità di tempo. Da questa sostituzione ho osservato i seguenti conteggi per ogni modello di sito:
| A | B | C | |
|---|---|---|---|
| A | 91 | 9 | 0 |
| B | 5 | 86 | 9 |
| C | 0 | 9 | 91 |
all’Interno di questo relativamente breve durata, non sembra come se qualcuno Un<>Csubstitutions si sono verificati. Tuttavia quando reran la simulazione per 100 unitsof tempo:
| A | B | C | |
|---|---|---|---|
| A | 55 | 35 | 10 |
| B | 29 | 36 | 35 |
| C | 20 | 36 | 44 |
Come si può vedere, molti caratteri “A” sono state sostituite con “C” e viceversa. Più in generale, sotto un modello di siti finiti sostituzioni multiple, la distribuzione dei conteggi del modello di sito diventa molto più piatta oltre ad aumentare semplicemente la proporzione di fuori diagonale rispetto ai conteggi diagonali.Le matrici di punteggio PAM e BLOSUM rappresentano più sostituzioni in modi radicalmente diversi.
Le matrici PAM per gli amminoacidi, insieme alle abbreviazioni a lettera singola utilizzate per gli amminoacidi geneticamente codificati, sono state sviluppate da MargaretDayhoff. Essi sono stati originariamente pubblicati nel 1978, e sulla base delle proteinsequences Dayhoff era stato compilando dal 1960, pubblicato come theAtlas di sequenza proteica e struttura.
Il nome PAM deriva da “point accepted mutation”, e si riferisce alla sostituzione di un singolo amminoacido in una proteina con un aminoacido diverso.Queste mutazioni sono state identificate confrontando sequenze altamente simili con almeno l ‘ 85% di identità, e si presume che eventuali sostituzioni osservate siano state il risultato di una singola mutazione tra la sequenza ancestrale e una delle sequenze del giorno presenti.
PAM definisce anche un’unità di tempo, dove 1 PAM è il tempo in cui ci si aspetta che 1/100 aminoacidi subiscano una mutazione. La matrice di probabilità PAM1 mostra la probabilità che l’amminoacido alla colonna j venga sostituito dall’amminoacido alla riga i. È stato calcolato dai conteggi PAM di Dayhoff e riscalato per 1 unità di tempo PAM. Come puoi vedere, le probabilità off-diagonali nella matrice PAM1 sono tutte molto piccole (tutti gli elementi sono stati scalati di 10.000 per la leggibilità):
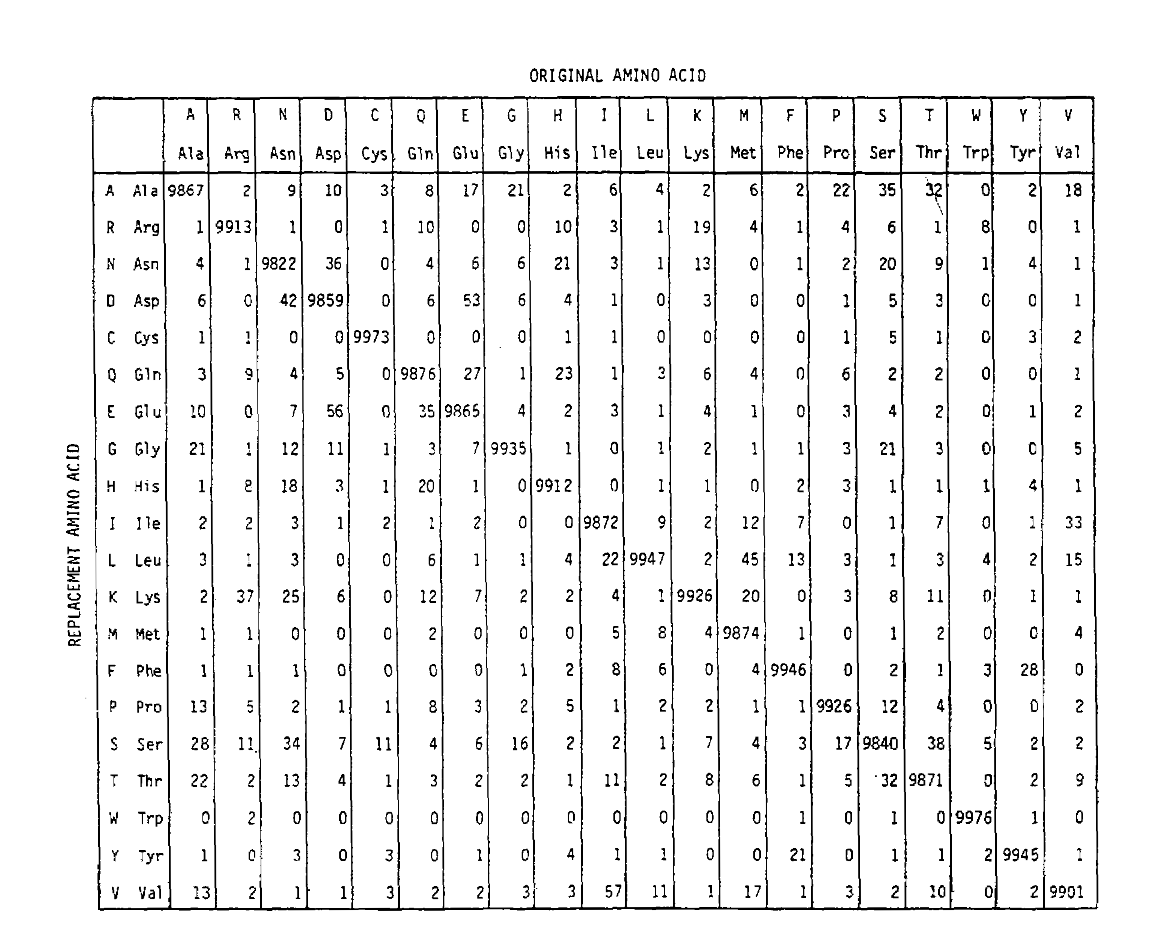
Per calcolare le probabilità di sostituzione degli amminoacidi per periodi più lunghi, la matrice può essere moltiplicata da sola il numero di volte corrispondente. Così la matrice di probabilità PAM250, descrivendo le probabilità di posizionamento date 250 unità di tempo PAM, è stata derivata aumentando la matrice di probabilità PAM1 alla potenza 250 (tutti gli elementi sono stati scalati di 100 per la leggibilità):
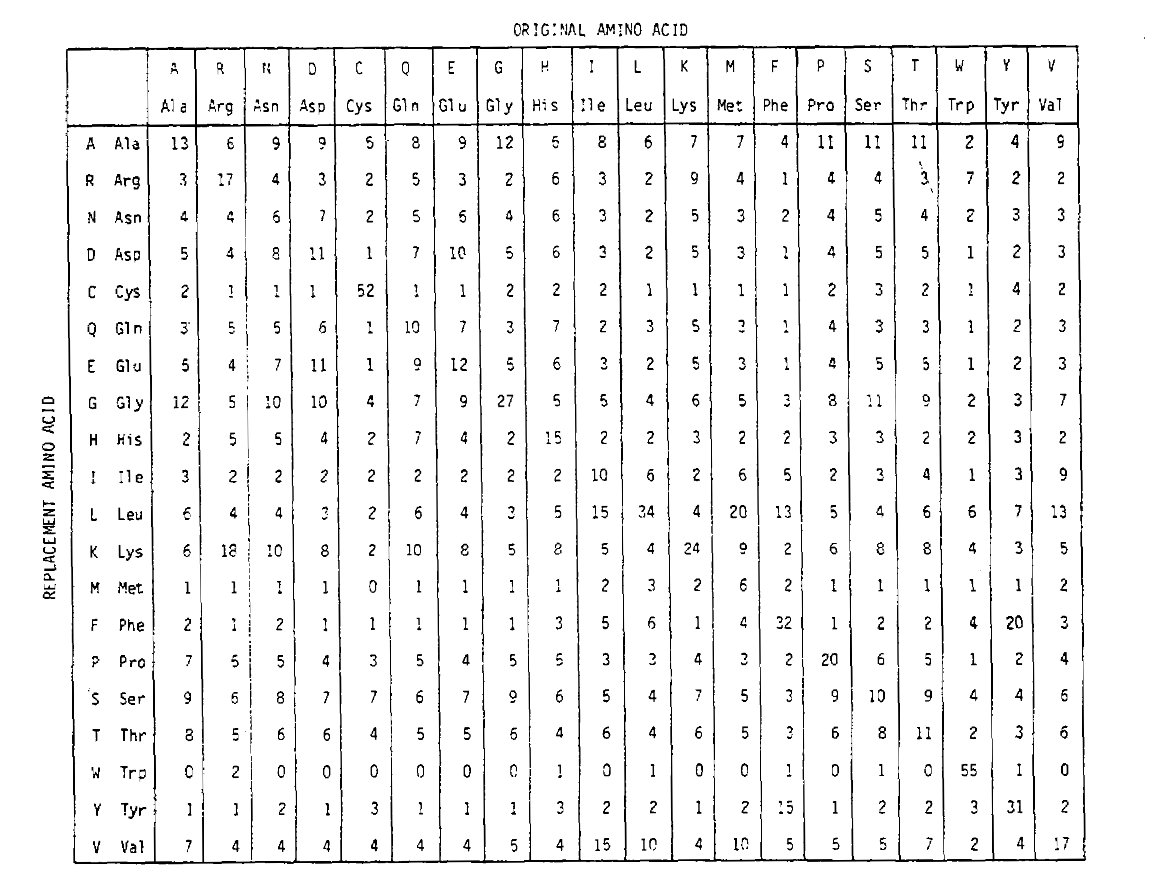
Le probabilità di sostituzione derivate usando questa esponenziazionecontengono correttamente per più sostituzioni. Non solo le off-diagonalprobabilities sono proporzionalmente più grandi come ci si aspetterebbe per una durata temporale più lunga, ma sono più piatte. Ad esempio, la probabilità di una sostituzione da valina (V)a isoleucina (I) è 33× più grande di una sostituzione da V a istadina (H)nella matrice PAM1, ma solo 4,5× più grande nella matrice PAM250.
Le matrici di punteggio possono quindi essere calcolate dalle matrici di probabilità e dalle frequenze di base osservate.
Le matrici BLOSUM, sviluppate da Steven e Jorja Henikoff e pubblicate nel 1992, hanno un approccio molto diverso. Mentre PAM applica implicitamente un modello stazionario di siti finiti di evoluzione utilizzando l’esponenziazione della matrice, l’effetto di sostituzioni multiple viene trattato implicitamente in BLOSUM strutturando matrici di punteggio diverse per diverse scale temporali.
All’interno di allineamenti di sequenze multiple di sequenze omologhe, vengono identificati blocchi contigui conservati di amminoacidi. All’interno di ogni blocco, multiplesequences sono raggruppati quando la loro identità di sequenza media a coppie è più alta di una certa soglia. La soglia è 80% per la matrice BLOSUM80, 62% per BLOSUM62, 50% per BLOSUM50 e così via.
Ciò significa che per BLOSUM80, i blocchi avranno un’identità media a coppie non superiore all ‘ 80%, per BLOSUM62 non superiore al 62%, eccetera.
Le probabilità di sostituzione degli amminoacidi per sequenze omologhe sono calcolate da confronti a coppie tra cluster. Queste probabilità saranno il risultato di sostituzioni singole e multiple, con sostituzioni multiple che avranno maggiore influenza a distanze evolutive maggiori. Pertanto, le scorematrices generate da confronti a coppie tra cluster di distanza media maggiore, come la matrice BLOSUM50, rappresenteranno naturalmente l’effetto più grande di sostituzioni multiple.
Anche se prendono percorsi diversi, le matrici di punteggio BLOSUM e PAM finali sono in realtà piuttosto simili. Secondo Henikoff e Henikoff, le seguenti matrici PAM e BLOSUM sono comparabili:
| PAM | BLOSUM |
|---|---|
| PAM250 | BLOSUM45 |
| PAM160 | BLOSUM62 |
| PAM120 | BLOSUM80 |
For more information on PAM (Dayhoff) and BLOSUM matrices, see capitolo 2 dianalisi biologica della sequenza di Durbin et al., e Wikipedia.
Aggiornamento 13 ottobre 2019: per un’altra prospettiva sulle matrici di sostituzione, consultare la sezione” Deviazioni ” alla fine del capitolo 5 degli algoritmi di bioinformatica (2a o 3a edizione) di Compeau e Pevzner.