doseringsvorm: injectie
medisch beoordeeld door Drugs.com. laatst bijgewerkt op 22 Dec 2020.
- bijwerkingen
- dosering
- professionele
- interacties
- zwangerschap
- meer
RX alleen.
steriele intraveneuze oplossing
- Inamrinon beschrijving
- Inamrinon – Klinische Farmacologie
- farmacokinetiek
- farmacodynamiek
- indicaties en gebruik voor Inamrinon
- contra-indicaties
- waarschuwingen
- voorzorgsmaatregelen
- Algemeen
- laboratoriumtesten
- geneesmiddelinteracties
- chemische interacties
- carcinogenese, mutagenese, verminderde vruchtbaarheid
- zwangerschap
- moeders die borstvoeding geven
- gebruik bij kinderen
- bijwerkingen
- behandeling van bijwerkingen
- overdosering
- Inamrinon dosering en toediening
- chemische interacties
- Hoe wordt Inamrinone geleverd
- VIAL LABEL
- More about inamrinone
- Related treatment guides
Inamrinon beschrijving
Inamrinon injectie USP vertegenwoordigt een nieuwe klasse van cardiale inotrope middelen die verschillen van digitalisglycosiden of catecholamines. Inamrinonlactaat wordt chemisch aangeduid als 5-Amino-6 (1H)-on 2-hydroxypropanaat en heeft de volgende structuur:
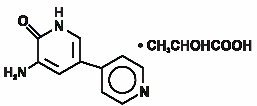
Inamrinon is een lichtgele kristallijne verbinding met een molecuulgewicht van 187,20 en een molecuulformule van C10H9N3O. elke mol melkzuur heeft een molecuulgewicht van 90,08 en een empirische formule van C3H6O3. De oplosbaarheden van inamrinone bij PH ‘ s 4.1, 6.0, en 8.0 zijn respectievelijk 25, 0,9, en 0,7 mg/mL.
Inamrinone injectie is een heldere gele steriele oplossing beschikbaar in 20 mL injectieflacons voor intraveneuze toediening. Elke mL bevat Inamrinonlactaat overeenkomend met 5 mg inamrinon en 0,25 mg natriummetabisulfiet toegevoegd als conserveermiddel in Water voor injectie. Alle doseringen in de bijsluiter worden uitgedrukt in termen van de base, Inamrinon. De pH wordt met melkzuur of natriumhydroxide ingesteld op 3,2 tot 4,0. De totale concentratie melkzuur kan variëren tussen 5 mg en 7,5 mg.
Inamrinon – Klinische Farmacologie
Inamrinon is een positief inotroop middel met vaatverwijdende activiteit, dat qua structuur en werkingswijze verschilt van digitalisglycosiden of catecholaminen.
Het mechanisme van de inotrope en vaatverwijdende effecten is niet volledig opgehelderd.
wat het inotrope effect betreft, wijzen experimentele gegevens erop dat het geen bèta-adrenerge agonist is. Het remt myocardiale cyclische adenosine monofosfaat (C-AMP) fosfodiësterase activiteit en verhoogt cellulaire niveaus van c-AMP. In tegenstelling tot digitalis, remt het de activiteit van natrium-kaliumadenosine trifosfatase niet.
met betrekking tot de vaatverwijdende activiteit vermindert Inamrinon de afterload en preload door het directe relaxerende effect op de gladde spieren van de bloedvaten.
farmacokinetiek
na intraveneuze bolusinjectie (1 tot 2 minuten) van 0,68 mg/kg tot 1,2 mg/kg bij normale vrijwilligers had Inamrinone een verdelingsvolume van 1,2 liter/kg en na een distributiefase halfwaardetijd van ongeveer 4,6 minuten in plasma, een gemiddelde schijnbare eerste-orde terminale eliminatiehalfwaardetijd van ongeveer 3,6 uur. Bij patiënten met congestief hartfalen die infusies met inamrinon kregen, was de gemiddelde schijnbare terminale eliminatiehalfwaardetijd van de eerste orde ongeveer 5,8 uur.
Inamrinon is in een studie in vitro 10% tot 22% gebonden aan humaan plasma-eiwit door ultrafiltratie, en in een andere studie 35% tot 49% gebonden door ultrafiltratie of evenwichtsdialyse.
de primaire excretieroute bij de mens is via de urine als zowel Inamrinon als verscheidene metabolieten (N-glycolyl, n-acetaat, O-glucuronide en N-glucuronide). Bij normale vrijwilligers werd ongeveer 63% van een orale dosis van 14C-gelabeld Inamrinon uitgescheiden in de urine gedurende een periode van 96 uur. In de eerste 8 uur was 51% van de radioactiviteit in de urine Inamrinon met 5% als n-acetaat, 8% als n-glycolaat en minder dan 5% voor elk glucuronide. Ongeveer 18% van de toegediende dosis werd binnen 72 uur in de ontlasting uitgescheiden.
in een 24-uurs nonradioactief intraveneus onderzoek werd 10% tot 40% van de dosis in de urine uitgescheiden als onveranderd Inamrinon, waarbij de n-acetylmetaboliet minder dan 2% van de dosis vertegenwoordigde.
bij patiënten met congestief hartfalen konden na een oplaadbolusdosis steady-state plasmaspiegels van ongeveer 2,4 mcg/mL worden gehandhaafd door een infusie van 5 mcg/kg/min tot 10 mcg/kg/min. Bij sommige patiënten met congestief hartfalen, met bijbehorende verminderde nier-en leverperfusie, is het mogelijk dat de plasmaspiegels van Inamrinon tijdens de infusieperiode stijgen; daarom kan het bij deze patiënten noodzakelijk zijn om de hemodynamische respons en/of geneesmiddelspiegel te controleren. De belangrijkste maatstaven voor de respons van de patiënt zijn de cardiale index, de pulmonale capillaire wigdruk, de centrale veneuze druk en hun relatie tot plasmaconcentraties. Bovendien, metingen van bloeddruk, urine output, en lichaamsgewicht kan nuttig blijken, zoals dergelijke klinische symptomen zoals orthopneu, dyspneu, en vermoeidheid.
farmacodynamiek
bij patiënten met een gedeprimeerde myocardiale functie geeft Inamrinon een onmiddellijke toename van het cardiale output als gevolg van zijn inotrope en vasodilaterende werking.
na een enkelvoudige intraveneuze bolusdosis Inamrinon van 0.75 mg / kg tot 3 mg/kg bij patiënten met congestief hartfalen treden dosisgerelateerde maximale verhogingen van het hartminuutvolume op (van ongeveer 28% bij 0,75 mg/kg tot ongeveer 61% bij 3 mg / kg). Het maximale effect treedt bij alle doses binnen 10 minuten op. De duur van het effect is afhankelijk van de dosis en duurt ongeveer 1/2 uur bij 0,75 mg/kg en ongeveer 2 uur bij 3 mg/kg.
bij dezelfde doses vertonen de pulmonale capillaire wigdruk en de totale perifere weerstand dosisafhankelijke dalingen (gemiddelde maximale dalingen van 29% in de pulmonale capillaire wigdruk en 29% in de systemische vasculaire weerstand). Bij doses tot 3 mg/kg zijn dosisafhankelijke dalingen van de diastolische druk (tot 13%) waargenomen. De gemiddelde arteriële druk neemt af (9,7%) bij een dosis van 3 mg/kg. De hartslag is over het algemeen onveranderd.
de veranderingen in hemodynamische parameters worden gehandhaafd tijdens continue intraveneuze infusie en gedurende enkele uren daarna.
Inamrinon is effectief bij volledig gedigitaliseerde patiënten zonder tekenen van cardiale glycosidetoxiciteit te veroorzaken. De inotrope effecten zijn additief aan die van digitalis. In gevallen van atriumflutter/fibrilleren is het mogelijk dat Inamrinon de ventrikelrespons kan verhogen vanwege de lichte verhoging van de A/V-geleiding. In deze gevallen wordt voorafgaande behandeling met digitalis aanbevolen.
verbetering van de linkerventrikelfunctie en verlichting van congestief hartfalen bij patiënten met ischemische hartziekten zijn waargenomen. De verbetering is opgetreden zonder symptomen of elektrocardiografische tekenen van myocardiale ischemie te induceren.
bij constante hartslag en bloeddruk treedt een toename van het cardiale output op zonder meetbare toename van het zuurstofverbruik van het myocard of veranderingen in arterioveneus zuurstofverschil.
inotrope activiteit wordt gehandhaafd na herhaalde intraveneuze doses Inamrinon. Toediening van inamrinon heeft hemodynamische en symptomatische voordelen voor patiënten die niet voldoende onder controle kunnen worden gehouden met conventionele therapie met diuretica en hartglycosiden.
indicaties en gebruik voor Inamrinon
Inamrinon injectie is voor de kortdurende behandeling van congestief hartfalen. Vanwege beperkte ervaring en de kans op ernstige bijwerkingen (zie bijwerkingen), mag Inamrinone alleen worden gebruikt bij patiënten die nauwkeurig kunnen worden gecontroleerd en die niet adequaat hebben gereageerd op digitalis, diuretica en/of vasodilatatoren. De ervaring met intraveneus Inamrinon in gecontroleerde onderzoeken duurt niet langer dan 48 uur na herhaalde bolussen en / of continue infusies.
of het nu oraal, continu intraveneus of intermitterend intraveneus wordt toegediend, noch Inamrinon, noch enig ander cyclisch-AMP-afhankelijk inotroop is in gecontroleerde studies aangetoond dat het veilig of effectief is bij de langdurige behandeling van congestief hartfalen. In gecontroleerde onderzoeken naar chronische orale therapie met verschillende van deze middelen (waaronder Inamrinon) werden de symptomen niet consistent verlicht en werden cyclisch-AMP-afhankelijke inotropen consistent geassocieerd met verhoogde risico ‘ s op ziekenhuisopname en overlijden. Patiënten met NYHA klasse IV symptomen bleken een bijzonder risico te lopen.
contra-indicaties
Inamrinon is gecontra-indiceerd bij patiënten die hiervoor overgevoelig zijn.
Het is ook gecontra-indiceerd bij patiënten waarvan bekend is dat ze overgevoelig zijn voor bisulfieten.
waarschuwingen
bevat natriummetabisulfiet, een sulfiet dat allergische reacties kan veroorzaken, waaronder anafylactische symptomen en levensbedreigende of minder ernstige astmatische episodes bij bepaalde gevoelige personen. De algemene prevalentie van sulfietgevoeligheid bij de algemene bevolking is onbekend en waarschijnlijk laag. Sulfietgevoeligheid wordt vaker gezien bij astmapatiënten dan bij niet-astmapatiënten.
voorzorgsmaatregelen
Algemeen
Inamrinone mag niet worden gebruikt bij patiënten met een ernstige aorta-of pulmonale valvulaire aandoening in plaats van chirurgische verlichting van de obstructie. Net als andere inotrope middelen, kan het de obstructie van het uitstroomkanaal in hypertrofische subaortic stenose verergeren.
tijdens intraveneuze therapie met inamrinon dienen de bloeddruk en de hartslag te worden gecontroleerd en dient de infusiesnelheid te worden vertraagd of gestopt bij patiënten die een excessieve daling van de bloeddruk vertonen.
patiënten die krachtige diuretische therapie hebben gekregen, kunnen onvoldoende hartvullende druk hebben om adequaat te reageren op Inamrinon, in welk geval voorzichtige liberalisering van de inname van vocht en elektrolyten geïndiceerd kan zijn.
supraventriculaire en ventriculaire aritmieën zijn waargenomen in de behandelde populatie met een zeer hoog risico. Hoewel niet is aangetoond dat Inamrinon op zich aritmogeen is, kan het potentieel voor aritmie, aanwezig bij congestief hartfalen zelf, worden verhoogd door elk geneesmiddel of combinatie van geneesmiddelen.
trombocytopenie en hepatotoxiciteit zijn waargenomen (zie bijwerkingen).
gebruik bij acuut myocardinfarct
er zijn geen klinische studies uitgevoerd bij patiënten in de acute fase van posthyocardinfarct. Daarom wordt in deze gevallen Inamrinon niet aanbevolen.
laboratoriumtesten
vocht en elektrolyten
veranderingen in vloeistof en elektrolyten en de nierfunctie moeten zorgvuldig worden gecontroleerd tijdens de behandeling met Inamrinon. Verbetering van het hartminuutvolume met als gevolg diurese kan een verlaging van de dosis diureticum noodzakelijk maken. Kaliumverlies als gevolg van overmatige diurese kan gedigitaliseerde patiënten predisponeren voor aritmieën. Daarom moet hypokaliëmie worden gecorrigeerd door kaliumsuppletie vóór of tijdens het gebruik van Inamrinon.
geneesmiddelinteracties
in een relatief beperkte ervaring zijn geen ongewenste klinische manifestaties waargenomen bij patiënten waarbij Inamrinon gelijktijdig werd gebruikt met de volgende geneesmiddelen: digitalisglycosiden; lidocaïne, kinidine; metoprolol, propranolol; hydralazine, prazosine; isosorbidedinitraat, nitroglycerine; chloortalidon, ethacrynzuur, furosemide, hydrochloorthiazide, spironolacton; captopril; heparine, warfarine; kaliumsupplementen; insuline; diazepam.
Eén geval van overmatige hypotensie is gemeld wanneer Inamrinon gelijktijdig met disopyramide werd gebruikt.
totdat meer ervaring beschikbaar is, dient gelijktijdige toediening met disopyramide met voorzichtigheid te worden uitgevoerd.
chemische interacties
een chemische interactie treedt langzaam op gedurende een periode van 24 uur wanneer de intraveneuze oplossing van Inamrinon direct wordt gemengd met dextrose (glucose) bevattende oplossingen. Daarom mag Inamrinon niet vóór injectie worden verdund met oplossingen die DEXTROSE (GLUCOSE) bevatten.
een chemische interactie treedt onmiddellijk op, hetgeen wordt aangetoond door de vorming van een precipitaat wanneer furosemide wordt geïnjecteerd in een intraveneuze lijn van een infusie van Inamrinon. Daarom dient furosemide niet te worden toegediend via intraveneuze lijnen die Inamrinon bevatten.
carcinogenese, mutagenese, verminderde vruchtbaarheid
Er was geen suggestie van een carcinogeen potentieel met Inamrinon bij orale toediening gedurende maximaal twee jaar aan ratten en muizen bij dosisniveaus tot de maximaal getolereerde dosis van 80 mg/kg/dag.
De micronucleustest bij muizen (bij 7,5 tot 10 maal de maximale dosis bij de mens) en de chromosoomafwijktest bij de Chinese hamsterovaria waren positief, wat zowel een clastogeen potentieel als een suppressie van het aantal polychromatische erytrocyten aangaf. Echter, de Ames Salmonella assay, muis lymfoom studie, en gekweekte menselijke lymfocyt metafase analyse waren allemaal negatief. De clastogene effecten zijn in tegenstelling tot de negatieve resultaten verkregen in de mannelijke en vrouwelijke vruchtbaarheidsstudies bij ratten, en een driegeneratiestudie bij ratten, beide met orale toediening.
bij doses van 50 mg/kg/dag en 100 mg/kg/dag werd een lichte verlenging van de drachtperiode bij ratten waargenomen. Dystokie trad op bij moederdieren die 100 mg/kg/dag kregen, wat resulteerde in een toename van het aantal doodgeborenen, een verminderde nestgrootte en een slechte overleving van de jongen.
zwangerschap
teratogene effecten-zwangerschapscategorie C
in Nieuw-Zeelandse witte konijnen bleek Inamrinon foetale skeletafwijkingen en grove uitwendige misvormingen te veroorzaken bij orale doses van 16 mg/kg en 50 mg / kg die toxisch waren voor het konijn. Studies bij Franse hy / Cr konijnen waarbij orale doses tot 32 mg/kg/dag werden gebruikt, bevestigden deze bevinding niet. Er werden geen misvormingen waargenomen bij ratten die intraveneus Inamrinon kregen toegediend in de maximale gebruikte dosis, 15 mg/kg/dag (ongeveer de aanbevolen dagelijkse intraveneuze dosis voor patiënten met congestief hartfalen). Er zijn geen adequate en goed gecontroleerde onderzoeken bij zwangere vrouwen uitgevoerd. Inamrinone mag alleen tijdens de zwangerschap worden gebruikt als het potentiële voordeel opweegt tegen het potentiële risico voor de foetus.
moeders die borstvoeding geven
voorzichtigheid is geboden wanneer Inamrinone wordt toegediend aan vrouwen die borstvoeding geven, omdat niet bekend is of het in de moedermelk wordt uitgescheiden.
gebruik bij kinderen
veiligheid en werkzaamheid bij pediatrische patiënten zijn niet vastgesteld.
bijwerkingen
trombocytopenie: Intraveneuze injectie van inamrinon leidde bij 2,4 procent van de patiënten tot een daling van het aantal bloedplaatjes tot minder dan 100.000/mm3 of tot normale limieten.
het komt vaker voor bij patiënten die langdurig worden behandeld. Tot op heden zijn in nauwkeurig gecontroleerde klinische studies bij patiënten bij wie het niet toegestaan was de bloedplaatjesaantallen laag te houden, geen bloedingsverschijnselen waargenomen.
Trombocytenreductie is dosisafhankelijk en treedt op als gevolg van een afname van de trombocytenoverlevingstijd. Verschillende patiënten die trombocytopenie ontwikkelden tijdens de behandeling met Inamrinon, ondergingen een beenmergonderzoek dat normaal was. Er is geen bewijs dat bloedplaatjesreductie verband houdt met de immuunrespons of met een bloedplaatjesactiverende factor.
gastro-intestinale effecten: gastro-intestinale bijwerkingen gemeld met Inamrinon tijdens klinisch gebruik waren misselijkheid (1,7%), braken (0,9%), buikpijn (0,4%) en anorexia (0,4%).
cardiovasculaire effecten: cardiovasculaire bijwerkingen die gemeld zijn met Inamrinon zijn onder meer aritmie (3%) en hypotensie (1,3%).
levertoxiciteit: Bij honden vertoonde Inamrinon bij IV-doses tussen 9 mg/kg/dag en 32 mg/kg/dag dosisgerelateerde hepatotoxiciteit die zich manifesteerde als enzyme elevation of hepatische celnecrose of beide. Hepatotoxiciteit is bij de mens waargenomen na langdurige orale toediening en is in beperkte mate (0,2%) waargenomen na intraveneuze toediening van Inamrinon. Er zijn ook zeldzame meldingen geweest van verhoging van enzymen en bilirubine en geelzucht.
overgevoeligheid: er zijn meldingen geweest van verschillende klaarblijkelijke overgevoeligheidsreacties bij patiënten die gedurende ongeveer twee weken werden behandeld met oraal Inamrinon. De klachten en symptomen waren variabel, maar omvatten pericarditis, pleuritis en ascites (1 geval), myositis met interstitiële shadowing op röntgenfoto ‘ s van de borst en verhoogde sedimentatiesnelheid (1 geval) en vasculitis met nodulaire pulmonale dichtheden, hypoxemie en geelzucht (1 geval). De eerste patiënt overleed, niet noodzakelijk aan de mogelijke reactie, terwijl de laatste twee verdwenen na het staken van de behandeling. Geen van de gevallen werd opnieuw aangekaart zodat de toewijzing aan Inamrinon niet zeker is, maar mogelijke overgevoeligheidsreacties MOETEN in overweging worden genomen bij elke patiënt die gedurende een langere periode Inamrinon gebruikt.
Algemeen: bijkomende bijwerkingen die zijn waargenomen in klinische onderzoeken met intraveneuze toediening van Inamrinone zijn koorts (0,9%), pijn op de borst (0,2%) en een branderig gevoel op de injectieplaats (0,2%).
behandeling van bijwerkingen
verlaging van het aantal bloedplaatjes: De asymptomatische verlaging van het aantal bloedplaatjes (tot <150.000/mm3) kan binnen een week na een verlaging van de geneesmiddeldosering worden teruggedraaid. Verder, zonder verandering in drugdosering, kan de telling op lager dan predrugniveaus zonder enige klinische gevolgen stabiliseren. Pre-drug bloedplaatjes tellingen en frequente bloedplaatjes tellingen tijdens de behandeling worden aanbevolen om te helpen bij beslissingen met betrekking tot dosisaanpassingen.
indien een trombocytenaantal van minder dan 150.000 / mm3 optreedt, kunnen de volgende acties worden overwogen: :
• De totale dagelijkse dosis onveranderd te houden, aangezien in sommige gevallen het aantal gestabiliseerd is of is teruggekeerd naar het niveau van vóór de behandeling. * Verlaag de totale dagelijkse dosis. * Stop met Inamrinone als, naar het klinisch oordeel van de arts, het risico groter is dan het potentiële voordeel.
gastro-intestinale bijwerkingen: hoewel gastro-intestinale bijwerkingen zelden werden waargenomen bij intraveneuze therapie, kan de arts, indien ernstige of verzwakkende bijwerkingen zouden optreden, de dosering willen verlagen of het geneesmiddel willen staken op basis van de gebruikelijke baten-risico-overwegingen.
levertoxiciteit: In klinische ervaring tot op heden met intraveneuze toediening is hepatotoxiciteit zelden waargenomen. Indien acute duidelijke veranderingen in leverenzymen optreden samen met klinische symptomen die wijzen op een idiosyncratische overgevoeligheidsreactie, dient de behandeling met Inamrinon onmiddellijk te worden gestaakt.
indien minder dan duidelijke enzymveranderingen optreden zonder klinische symptomen, dienen deze niet-specifieke veranderingen op individuele basis te worden geëvalueerd. De arts kan Inamrinone willen voortzetten, de dosering willen verlagen of het geneesmiddel willen staken op basis van de gebruikelijke voordelen/risico-overwegingen.
overdosering
overlijden is gemeld bij een massale accidentele overdosering (840 mg gedurende drie uur door initiële bolus en infusie) van Inamrinon, hoewel een causaal verband onzeker is. Tijdens de bereiding en toediening van het product dient zorgvuldigheid te worden betracht.
Doses Inamrinon kunnen hypotensie veroorzaken vanwege het vaatverwijdende effect. Als dit gebeurt, moet de toediening van Inamrinon worden verminderd of stopgezet. Er is geen specifiek antidotum bekend, maar algemene maatregelen voor ondersteuning van de bloedsomloop moeten worden genomen.
bij ratten was de LD50 van inamrinon, als lactaatzout, intraveneus 102 mg/kg of 130 mg/kg in twee verschillende studies en 132 mg/kg oraal (intragastrisch); als suspensie in waterige tragacanth was de orale LD50 239 mg/kg.
Inamrinon dosering en toediening
Oplaaddoses van inamrinon injectie dienen te worden toegediend zoals geleverd (onverdund). Infusies van Inamrinone kunnen worden toegediend in een normale of half normale zoutoplossing tot een concentratie van 1 mg/mL tot 3 mg/mL. Verdunde oplossingen moeten binnen 24 uur worden gebruikt.
Inamrinon injectie kan worden toegediend in lopende dextrose (glucose) infusies via een Y-Connector of rechtstreeks in de slang waar de voorkeur wordt gegeven.
chemische interacties
een chemische interactie treedt langzaam op gedurende een periode van 24 uur wanneer de intraveneuze oplossing van Inamrinon direct wordt gemengd met dextrose (glucose) bevattende oplossingen. Daarom mag Inamrinon niet vóór injectie worden verdund met oplossingen die DEXTROSE (GLUCOSE) bevatten.
een chemische interactie treedt onmiddellijk op, hetgeen wordt aangetoond door de vorming van een precipitaat wanneer furosemide wordt geïnjecteerd in een intraveneuze lijn van een infusie van Inamrinon. Daarom dient furosemide niet te worden toegediend via intraveneuze lijnen die Inamrinon bevatten.
de volgende procedure wordt aanbevolen voor de toediening van inamrinone injectie:
1. Start de behandeling met een oplaaddosis van 0,75 mg/kg die langzaam wordt toegediend gedurende 2 tot 3 minuten.
|
LOADING DOSE DETERMINATION 0.75 mg/kg (undiluted) |
||||||||||
|
Patient Weight in kg |
||||||||||
|
mL of undiluted Inamrinone Injection |
||||||||||
2. Continue therapy with a maintenance infusion between 5 mcg/kg/min and 10 mcg/kg/min.
3. Based on clinical response, an additional loading dose of 0.75 mg/kg may be given 30 minutes after the initiation of therapy.
4. De infusiesnelheid varieert gewoonlijk van 5 mcg/kg/min tot 10 mcg/kg/min, zodat de aanbevolen totale dagelijkse dosis (inclusief oplaaddoses) niet hoger is dan 10 mg / kg. Een beperkt aantal patiënten die met hogere doses zijn onderzocht, ondersteunt een doseringsschema tot 18 mg/kg / dag voor een verkorte duur van de behandeling.
de volgende infusiesnelheidsgrafiek kan worden gebruikt om te verzekeren dat de berekeningen correct zijn uitgevoerd.
om de grafiek te gebruiken, moet de concentratie van de gebruikte inamrinon-oplossing voor infusie 2,5 mg/mL (2500 mcg/mL) zijn. Deze concentratie wordt bereid door de inamrinonoplossing te mengen met een gelijk volume oplosmiddel (normale of half normale zoutoplossing).
| * verdunning: voor de bereiding van de 2,5 mg / mL concentratie aanbevolen voor infusie mix Inamrinon met een gelijk volume verdunningsmiddel. Meng bijvoorbeeld drie injectieflacons van 20 mL Inamrinone (3 x 20 mL = 60 mL) met 60 mL verdunningsmiddel voor een totaal volume van 120 mL van de uiteindelijke 2,5 mg/mL oplossing van inamrinone. | ||||||||||
|
Inamrinone IV INFUSION RATE (mL/hr) CHART Using 2.5 mg/mL Infusion Concentration* |
||||||||||
|
Patient Weight in kg |
||||||||||
|
Dosage: 5.0 mcg/kg/min |
||||||||||
|
7.5 mcg/kg/min |
||||||||||
|
10.0 mcg/kg/min |
||||||||||
Example: A 70 kg patient would require a loading dose of 10.5 mL of undiluted Inamrinone. If the physician selects a dose of 7.5 mcg/kg/min voor de infusie zou het debiet 13 mL/uur zijn bij de 2,5 mg / mL concentratie Inamrinon.
5. De toedieningssnelheid en de duur van de behandeling moeten worden aangepast aan de respons van de patiënt. De arts kan de infusie verlagen of naar beneden titreren op basis van klinische respons of ongunstige effecten.
het bovenstaande doseringsschema plaatst de plasmaconcentratie van Inamrinon van de meeste patiënten op ongeveer 3 mcg / mL. Stijgingen van de cardiale index tonen een lineair verband met de plasmaconcentratie van 0.5 mcg / mL tot 7 mcg / mL. Er zijn geen waarnemingen gedaan bij hogere plasmaconcentraties.
verbetering van de patiënt kan worden weerspiegeld door een toename van het hartminuutvolume, een vermindering van de pulmonale capillaire wigdruk en dergelijke klinische reacties zoals een vermindering van dyspneu en een verbetering van andere symptomen van hartfalen, zoals orthopneu en vermoeidheid.
controle van de centrale veneuze druk (CVP) kan nuttig zijn bij de beoordeling van hypotensie en vochtbalansbeheersing. Voorafgaande correctie of aanpassing van vloeistof/elektrolyten is essentieel om een bevredigende respons met Inamrinon te verkrijgen.
parenterale geneesmiddelen moeten visueel worden geïnspecteerd en mogen niet worden gebruikt als deeltjes of verkleuring worden waargenomen.
Hoe wordt Inamrinone geleverd
Inamrinone injectie USP wordt geleverd in flacons met een enkelvoudige dosis van 20 mL steriele, heldere gele oplossing afzonderlijk verpakt. NDC 55390-042-10.
elke 1 mL bevat Inamrinonlactaat overeenkomend met 5 mg inamrinon.
beschermen tegen licht. De verpakking is lichtbestendig voor bescherming tijdens opslag. Bewaar in de doos tot het moment van gebruik.
Bewaren bij gecontroleerde kamertemperatuur 15 ° tot 30 ° C.
Manufactured by: Manufactured for:
Ben Venue Laboratories, Inc. Bedford Laboratories™
Bedford, OH 44146 Bedford, OH 44146
August 2002 AMR-P01
VIAL LABEL

Vial Label 100 mg/20 mL
| Inamrinone Inamrinone injection |
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
Labeler – Bedford Laboratories (884528407)
Registrant – Ben Venue Laboratories (004327953)
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Ben Venue Laboratories Inc. | 004327953 | MANUFACTURE(55390-042) | |
More about inamrinone
- Side Effects
- During Pregnancy
- Dosage Information
- Drug Interactions
- Drug class: inotropic agents
Related treatment guides
- Heart Failure
Medical Disclaimer