Het doel van deze tutorial is om u kennis te laten maken met de verwerking van de volgende generatie sequencing data in Galaxy. Deze tutorial maakt gebruik van een covid-19 variant bellen van Illumina data, maar het gaat niet over variant bellen per se.
aan het einde van deze tutorial zult u weten:
- Hoe gegevens te vinden in SRA en deze informatie over te dragen naar Galaxy
- Hoe basis NGS-gegevensverwerking uit te voeren in Galaxy inclusief:
- Quality Control (QC) van Illumina gegevens
- Mapping
- het Verwijderen van duplicaten
- Variant bellen met
lofreq - Variant annotatie
- met Behulp van de datasets collecties
- gegevens Importeren naar Jupyter
### Agenda>> In deze tutorial behandelen we:>> – 1. TOC> {: toc}>{: .agenda} # # twee paden door deze tutorialWe hebben twee trajecten gemaakt die je via deze tutorial kunt volgen.1. ** Traject 1 * * – start met NCBI ‘ s SRA en zoek naar beschikbare toetredingen → Start (#the-sequence-read-archive)2. ** Trajectory 2 * * – omzeil NCBI ‘ s SRA en begin direct met Galaxy. → Start (#back-in-galaxy) we raden aan te beginnen met **Traject 2**.# The Sequence Read ArchiveThe (https://www.ncbi.nlm.nih.gov/sra) is het primaire archief van *niet-geassembleerde reads* voor de (https://www.ncbi.nlm.nih.gov/). SRA is een geweldige plek om de sequencing data die ten grondslag liggen aan publicaties en studies te krijgen.Deze tutorial behandelt hoe u sequentiegegevens van SRA naar Galaxy kunt krijgen met behulp van een directe verbinding tussen de twee.> ### comment Comment>> U hoort ook dat SRA wordt aangeduid als het *short Read Archive*, de oorspronkelijke naam.> {:.commentaar} # # toegang tot SRASRA kan rechtstreeks worden bereikt via de website, of via het tool panel op Galaxy.> ### comment Comment>> aanvankelijk bestaat de tool panel optie voor toegang tot SRA alleen op de (https://usegalaxy.org/). Ondersteuning voor de directe verbinding met Sra zal worden opgenomen in de 20.05 release van Galaxy {:.commentaar}> # # # hands_on Hands-on: Explore Sra Entrez>> 1. Ga naar uw Galaxy-exemplaar naar keuze, zoals een van de (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) of een andere. (Deze tutorial gebruikt usegalaxy.org).> 1. Als uw geschiedenis nog niet leeg is, start dan een nieuwe geschiedenis (zie (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) voor meer informatie over Galaxy-geschiedenissen)> 1. ** Klik op* * `Gegevens ophalen` bovenaan het gereedschapsvenster.> 1. ** Klik op* * ‘Sra Server `in de lijst met tools weergegeven onder`Data ophalen’.> dit neemt u de (https://www.ncbi.nlm.nih.gov/sra) — U kunt ook direct starten vanuit de SRA. Een zoekvak wordt weergegeven aan de bovenkant van de pagina. Probeer te zoeken naar iets waarin je geà nteresseerd bent, zoals `dolphin` of `kidney` of `dolphin kidney` en klik vervolgens **op** de `Search` knop.>> dit geeft een lijst van *Sra experimenten* terug die overeenkomen met uw zoekopdracht. Sra-experimenten, ook bekend als *SRX-ingangen*, bevatten sequentiegegevens van een bepaald experiment, evenals een verklaring van het experiment zelf en andere gerelateerde gegevens. U kunt de geretourneerde experimenten te verkennen door te klikken op hun naam. Zie (https://www.ncbi.nlm.nih.gov/books / NBK56913/) in de (https://www.ncbi.nlm.nih.gov/books/n/helpsrakb/) voor meer informatie.>> wanneer u tekst invoert in het SRA zoekvak, gebruikt u (https://www.ncbi.nlm.nih.gov/sra/docs/srasearch/). Entrez ondersteunt zowel eenvoudige tekst zoekopdrachten, en zeer nauwkeurige zoekopdrachten die specifieke metadata controleren en willekeurig complexe logische uitdrukkingen gebruiken. Met Entrez kunt u uw zoekopdrachten opschalen van basic naar advanced Als u uw zoekopdrachten verkleint. De syntaxis van geavanceerde zoekopdrachten kan ontmoedigend lijken, maar SRA biedt een grafische (https://www.ncbi.nlm.nih.gov/sra/advanced/) om de specifieke syntaxis te genereren. En, zoals we hieronder zullen zien, de SRA Run Selector biedt een nog vriendelijker gebruikersinterface voor het vernauwen van onze geselecteerde gegevens.>> speel rond met de SRA Entrez-interface, inclusief de advanced query builder, om te zien of u een set SRA-experimenten kunt identificeren die relevant zijn voor een van uw onderzoeksgebieden.{: .hands_on}> # # # hands_on Hands-on: Genereer een lijst met overeenkomende experimenten met Entrez>> nu je een basiskennis hebt van Sra Entrez, laten we de sequenties vinden die in deze tutorial worden gebruikt.>> 1. Als u er nog niet bent, navigeer **terug naar (https://www.ncbi.nlm.nih.gov/sra> 1. ** Wis * * alle zoektekst uit het zoekvak.> 1. ** Typ* * `sars-cov-2 `in het zoekvak en **klik op**`Zoeken’.> dit geeft een lange lijst van SRA-experimenten terug die overeenkomen met onze zoekopdracht, en die lijst is veel te lang om te gebruiken in een tutorial-oefening. Op dit punt konden we gebruik maken van de geavanceerde Entrez query builder we geleerd over boven.> maar dat doen we niet. laten we in plaats daarvan de *too long for a tutorial* list results die we hebben naar de SRA Run Selector sturen, en de vriendelijker interface gebruiken om onze resultaten te verfijnen.>> !(../../ images / sra_entrez.png) {:.hands_on}> # # # hands_on Hands-on: Ga van Entrez naar Sra Run Selector>> Bekijk resultaten als een uitgebreide interactieve tabel met behulp van de RunSelector.>> 1. Klik op resultaten Verzenden om selector uit te voeren, die wordt weergegeven in een vak aan de bovenkant van de zoekresultaten.>> !(../../ images / sra_entrez_result.png)>>> # # # tip wat als u de Selectorkoppeling uitvoeren niet ziet?>>>> U kunt deze tekst eerder hebben opgemerkt toen u Entrez search aan het verkennen was. Deze tekst verschijnt slechts een deel van de tijd, wanneer het aantal zoekresultaten valt binnen een vrij breed venster. Je zult het niet zien als je maar een paar resultaten hebt, en je zult het niet zien als je meer resultaten hebt dan de Run Selector kan accepteren.>>>> *U moet Selector uitvoeren om uw resultaten naar Galaxy te sturen.* Wat als je niet genoeg resultaten hebt om deze link te activeren wordt getoond? In dat geval bel je get naar de Run Selector door **te klikken op** op de `Send to` pulldown menu in de rechterbovenhoek van het resultatenpaneel. Om Selector uit te voeren, **selecteer** `Run Selector` en vervolgens **klik op** de `Go` knop.> !(../../ images / sra_entrez_send_to.png)> {: .tip}>>> 1. ** Klik op* * `Send results to Run selector` bovenaan het zoekresultaten Paneel. (Als u deze link niet ziet, zie dan het commentaar direct hierboven.){: .hands_on} # # Sra Run SelectorWe hebben al eerder geleerd hoe we onze zoekresultaten kunnen verfijnen door de geavanceerde syntaxis van Entrez te gebruiken. Maar we maakten geen gebruik van die macht toen we in Entrez waren. In plaats daarvan hebben we een eenvoudige zoekopdracht gebruikt en vervolgens alle resultaten naar de Run Selector gestuurd. We hebben nog niet de (korte) lijst met resultaten die we willen analyseren. * What are we doing?* We gebruiken Entrez en de Run Selector hoe ze zijn ontworpen om te worden gebruikt: * gebruik de Entrez interface om uw resultaten te beperken tot een grootte die de Run Selector kan consumeren. * Stuur die Entrez resultaten naar de SRA Run Selector * gebruik de veel vriendelijkere interface van de Run Selector naar 1. Beter begrijpen van de gegevens die we hebben 1. Beperk die resultaten met die kennis.> ### comment Run Selector is zowel meer als minder dan Entrez>> Run Selector kan het meeste doen, maar niet alles wat Entrez search syntaxis kan doen. Run selector maakt gebruik van* faceted search * technologie die eenvoudig te gebruiken en krachtig is, maar die inherente grenzen heeft. Specifiek, Entrez zal beter werken bij het zoeken op attributen die tientallen, honderden, of duizenden verschillende waarden hebben. Run Selector zal beter werken zoeken attributen met minder dan 20 verschillende waarden. Gelukkig, dat beschrijft de meeste zoekopdrachten.{: .opmerking}Het Selectorvenster uitvoeren is verdeeld in verschillende Panelen:* **`Filters List`**: in de linkerbovenhoek. Hier zullen we onze zoektocht verfijnen.* **’Select’**: een samenvatting van wat in eerste instantie werd doorgegeven aan Selector uitvoeren, en hoeveel daarvan we tot nu toe hebben geselecteerd. (En tot nu toe hebben we er niets van geselecteerd.) Let ook op de verleidelijke, maar nog steeds grijze, `Galaxy` knop.** * ` Gevonden x Items ‘ * * aanvankelijk is dit de lijst van items die vanuit Entrez worden verzonden om Selector uit te voeren. Deze lijst zal krimpen als we er filters op Toepassen.!(../../images / sra_run_selector.png)> ### commentaar Waarom is het aantal gevonden items *gestegen?* >> herinneren dat de Entrez interface lijsten SRA experimenten (SRX entries). Voer Selector lists *runs* — sequencing datasets-en er zijn *een of meer* runs per experiment. We hebben dezelfde gegevens als voorheen, we zien het nu in fijnere details.{: .opmerking}de` Filters lijst ‘ in de linkerbovenhoek toont kolommen in onze resultaten die ofwel continue numerieke waarden, of 10 of minder (u kunt dit aantal wijzigen) verschillende waarden in hen. ** Scroll * * naar beneden door de lijst selecteer een paar van de filters. Wanneer een filter is geselecteerd, verschijnt een* waarden * box hieronder, met opties voor dit filter, en het aantal runs met elke optie. Deze waarden / opties worden getrokken uit de metadata van de dataset. Probeer **te selecteren** een paar interessante klinkende filters en vervolgens** selecteer * * een of meer opties voor elk filter. Probeer** deselecteren * * opties en filters. Terwijl u dit doet, zal het aantal gevonden resultaten afnemen of toenemen.> # # # tip Tip: Gebruik Filters om de gegevens beter te begrijpen>> Filters zijn hoe u de datasets die worden overwogen voor het verzenden naar Galaxy versmalt, maar ze zijn ook een uitstekende manier om uw gegevens te begrijpen:> eerst is het selecteren van een filter een gemakkelijke manier om het bereik van waarden in een kolom te zien. U kunt het misschien niet (https://www.google.com/search?q=sra+sirs_outcome), maar u kunt het mogelijk achterhalen door te zien welke waarden erin zitten.> ten tweede kunt u onderzoeken hoe verschillende kolommen zich tot elkaar verhouden. Is er een verband tussen `sirs_outcome` – waarden en `disease_stage` – waarden?{: .tip}> # # # hands_on Hands-on: Beperk uw resultaten met behulp van Run Selector>> 1. Als je filters hebt ingeschakeld, **deselecteer** ze.> zodra u dit hebt gedaan, zullen er geen *waarden* vakken verschijnen onder de `Filters lijst`.> 2. ** Kopieer en plak * * deze zoekstring in het zoekvak` gevonden Items’.> SRR11772204 OR SRR11597145 OR SRR11667145> deze handgekozen reeks runs beperkt onze resultaten tot 3 runs van verschillende geografische distributie.{: .hands_on}dit vermindert uw ‘Gevonden Items’ lijst van tienduizenden runs naar 3 runs (een beheersbaar aantal voor een tutorial!). Maar we zijn nog niet helemaal klaar met Run Selector. Merk op dat de ‘Galaxy’ knop nog steeds grijs is. We hebben onze opties verkleind, maar we hebben nog niets geselecteerd om naar Galaxy te sturen.Het is mogelijk om elke resterende run te selecteren door** te klikken op * * het vinkje aan de bovenkant van de eerste kolom. U kunt Alles deselecteren door * * te klikken op * * de ‘X’.> # # # hands_on Hands-on: selecteer Uitvoeren en stuur naar Galaxy>> 1. Selecteer alle runs door * * te klikken op * * de `X’.> en nu is de knop ‘Galaxy’ live.> 1. ** Klik op * * de `Galaxy` knop in de `Select` sectie aan de bovenkant van de pagina.{: .hands_on}# # terug in Galaxywanneer we op `Galaxy` klikken in Run Selector gebeuren er verschillende dingen. Eerste, het lanceert een nieuw tabblad browser of venster dat wordt geopend in Galaxy. U zult de * big green box* zien die aangeeft dat de handdruk tussen SRA en Galaxy succesvol was en u zult dan een nieuwe `SRA` – taak zien in uw geschiedenispaneel. Dit vak kan beginnen als grijs / in behandeling, wat aangeeft dat de overdracht nog niet is begonnen, of het kan rechtstreeks naar geel / lopend of naar groen / gedaan gaan.> # # # hands_on Hands-on: Onderzoek de nieuwe Sra-gegevensset>> 1. Zodra de` SRA ‘ -overdracht is voltooid, **klik** op het pictogram galaxy-eye (eye) van de dataset.>> dit toont de dataset in het middenpaneel van de Melkweg.{: .hands_on}de` SRA ‘ dataset is geen sequentiegegevens, maar eerder *metadata* die we zullen gebruiken om sequentiegegevens van SRA te krijgen. Deze metadata weerspiegelt de informatie die we zagen in de sectie `gevonden Items ‘ van de Run Selector. De metadata is niet de eindgegevens die we zoeken van SRA, maar het hebben van al die metadata is vaak nuttig in latere analysestappen.Laten we nu die metadata gebruiken om de sequentiegegevens van SRA op te halen. SRA biedt tools voor het extraheren van allerlei informatie, inclusief de sequentiegegevens zelf. De Galaxy Tool ‘Faster Download and Extract Reads in FASTQ’ is gebaseerd op de SRA (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump) utility, en doet precies dat.–>
- vind de benodigde gegevens in SRA
- hands_on Hands-on: taakbeschrijving
- commentaar commentaar
- proces en filter SraRunInfo.csv-bestand in Galaxy
- hands_on Hands-on: Upload SraRunInfo.csv-bestand in Galaxy
- opmerking pas op voor bezuinigingen
- hands_on Hands-on: het maken van een subset van gegevens
- tip Tip: Het vinden van tools
- download sequencing data with Faster Download and Extract Reads in FASTQ
- hands_on Hands-on: taakbeschrijving
- wat nu?
- Variatieanalyse van SARS-Cov-2 sequentiegegevens
- commentaar op de usegalaxy.* Covid-19 analyse project
- haal de referentie-genoomgegevens
- hands_on Hands-on: Get the reference genome data
- Tip: Importeren via links
- adapter trimmen met fastp
- hands_on Hands-on: Task description
- Uitlijning met de Kaart met de BWA-MEM
- hands_on Hands-on: Sequencing reads uitlijnen naar referentie-genoom
- duplicaten verwijderen met Markduplicaten
- hands_on Hands-on: Verwijder PCR duplicaten
- genereer uitlijningsstatistieken met samtools stats
- hands_on Hands-on: Genereren uitlijning statistieken
- Realign reads met lofreq viterbi
- hands_on Hands-on: Realign reads around indels
- voeg indel-kwaliteiten toe met lofreq voeg indel-kwaliteiten
- hands_on Hands-on: Toevoegen indel kwaliteiten
- Oproep Varianten met lofreq Oproep varianten
- hands_on Hands-on: Bel varianten
- annoteer variante effecten met SnpEff eff:
- hands_on Hands-on: annoteer varianteffecten
- Creëer een tabel met varianten met behulp van Snpsift-Extractvelden
- hands_on Hands-on: Create table of variants
- vat gegevens samen met MultiQC
- hands_on Hands-on: Resumize data
- Conclusie
- toetspunten sleutelpunten
- Veelgestelde vragen
- nuttige literatuur
- Feedback
- Citing this Tutorial
- details BibTeX
vind de benodigde gegevens in SRA
eerst moeten we een goede dataset vinden om mee te spelen. Het Sequence Read Archive (SRA) is het primaire archief van niet-geassembleerde leest beheerd door de Amerikaanse National Institutes of Health (NIH). SRA is een geweldige plek om de sequencing data die ten grondslag liggen aan publicaties en studies te krijgen. Laten we dat doen:
hands_on Hands-on: taakbeschrijving
- ga naar NCBI ’s SRA-pagina door uw browser naar https://www.ncbi.nlm.nih.gov/sra
- in het zoekveld voer
SARS-CoV-2 Patient Sequencing From Partners / MGH(U kunt ook gewoon op deze link klikken)
- de webpagina toont een groot aantal SRA-datasets (op het moment van schrijven waren er 2.223). Dit zijn gegevens uit een studie die de analyse beschrijft van SARS-CoV-2 in Boston en omgeving.
- Download metadata die deze datasets beschrijven door:
- klikken op Verzenden naar: dropdown
File- Formaat wijzigen naar
RunInfo- klikken op Bestand maken er zo uit zou moeten zien:
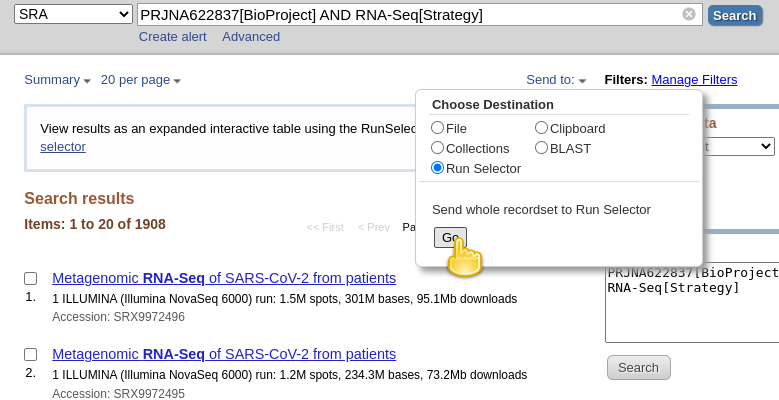
- Dit zou een vrij grote
SraRunInfo.csvbestand in uwDownloadsmap aanmaken.
nu we dit bestand hebben gedownload, kunnen we naar een Galaxy-instantie gaan en het verwerken.
commentaar commentaar
merk op dat het bestand dat we zojuist hebben gedownload geen sequentiegegevens zelf bevat. Eerder, is het metadata die eigenschappen beschrijven van het rangschikken leest. We zullen deze lijst filteren tot slechts een paar toetredingen die zullen worden gebruikt in de rest van deze tutorial.
proces en filter SraRunInfo.csv-bestand in Galaxy
hands_on Hands-on: Upload SraRunInfo.csv-bestand in Galaxy
- ga naar uw Galaxy-exemplaar naar keuze, zoals een van de usegalaxy.org, usegalaxy.eu, usegalaxy.org.au of een ander. (Deze tutorial gebruikt usegalaxy.org).
- klik op de knop Gegevens uploaden:
- klik in het dialoogvenster dat verschijnt op “Choose local files” knop:

- Zoek en selecteer
SraRunInfo.csvbestand vanaf uw computer- klik op startknop
- sluit het dialoogvenster door op knop Sluiten
- u kunt nu de inhoud van dit bestand bekijken door op het galaxy-eye (eye) – pictogram te klikken. U zult zien dat dit bestand veel informatie bevat over individuele SRA-toetredingen. In deze studie komt elke toetreding overeen met een individuele patiënt wiens monsters werden gesequenced.
Galaxy kan alle 2000+ datasets verwerken, maar om deze tutorial draaglijk te maken, moeten we een kleinere subset selecteren. In het bijzonder onze eerdere ervaring met deze gegevens toont twee interessante datasets SRR11954102 en SRR12733957. Laten we ze eruit trekken.
opmerking pas op voor bezuinigingen
de onderstaande Hands-on sectie gebruikt snijgereedschap. Er zijn twee snijgereedschappen in de Melkweg als gevolg van historische redenen. Dit voorbeeld gebruikt gereedschap met de volledige naam kolommen uit een tabel knippen (knippen). Echter, dezelfde logica geldt voor de andere tool. Het heeft gewoon een iets andere interface.
hands_on Hands-on: het maken van een subset van gegevens
- zoeken tool” selecteer Regels die overeenkomen met een expressie ” Tool in Filter en sorteer sectie van het gereedschapsvenster.
tip Tip: Het vinden van tools
Galaxy kan een overweldigende hoeveelheid tools hebben geïnstalleerd. Om een specifiek gereedschap te vinden, typt u de naam van het gereedschap in het zoekvak van het gereedschapspaneel om het gereedschap te vinden.
- zorg ervoor dat de
SraRunInfo.csvdataset die we zojuist hebben geüpload wordt weergegeven in het param-bestand” select lines from ” veld van het gereedschapsformulier.- voer in het veld” the pattern”de volgende uitdrukking in →
SRR12733957|SRR11954102. Dit zijn twee toevoegingen die we gescheiden willen vinden door het pipe symbool|. De|betekentor: zoek regels metSRR12733957ofSRR11954102.- klik op
Executeknop.- dit genereert een bestand met twee regels (goed … een regel wordt ook gebruikt als de header, dus het zal lijken dat het bestand drie regels heeft. Het is oké.)
- knip de eerste kolom uit het bestand met behulp van het gereedschap “knippen” gereedschap, dat u zult vinden in de sectie Tekstmanipulatie van het gereedschapsvenster.
- zorg ervoor dat de dataset die door de vorige stap is geproduceerd, is geselecteerd in het veld “file to cut” van het gereedschapsformulier.
- verander “Delimited by”in
Comma- In” List of fields”selecteer
Column: 1.- Hit
Executedit zal een tekstbestand met slechts twee regels produceren:SRR12733957SRR11954102
nu we identifiers van datasets hebben die we willen, moeten we de werkelijke sequencing data.
download sequencing data with Faster Download and Extract Reads in FASTQ
hands_on Hands-on: taakbeschrijving
- sneller download en Extract Reads in fastq tool met de volgende parameters:
- “select input type”:
List of SRA accession, one per line
- De parameter param-file” SRA toetredingslijst “moet wijzen op de uitvoer van het gereedschap” Cut ” uit de vorige stap.
- klik op de knop
Execute. Dit zal de tool draaien, die de reeks gelezen datasets ophaalt voor de runs die vermeld werden in deSRAdataset. Het kan even duren. Dus dit kan een goed moment zijn om koffie te halen.- verschillende items worden aangemaakt in uw geschiedenispaneel wanneer u deze taak verzendt:
Pair-end data (fasterq-dump): Bevat Paired-einde toevoegen (indien beschikbaar)Single-end data (fasterq-dump)Bevat Één-einde toevoegen (indien beschikbaar)Other data (fasterq-dump)Bevat een Ongepaard datasets (indien beschikbaar)fasterq-dump logBevat Informatie over het hulpprogramma uitvoeren
De eerste drie items zijn eigenlijk verzamelingen van datasets. Collecties in Galaxy zijn logische groepen van datasets die de semantische relaties tussen hen in het experiment / analyse weerspiegelen. In dit geval maakt de tool een aparte verzameling elk voor gepaarde-end leest, enkele leest, en andere.Zie de collecties tutorials voor meer informatie.
verken de collecties door eerst op de collectienaam te klikken in het geschiedenispaneel. Dit neemt je mee in de collectie en toont je de datasets erin. U kunt dan terug navigeren naar het buitenste niveau van uw geschiedenis.
zodra fasterq klaar is met het overbrengen van gegevens (alle vakken zijn groen / klaar), zijn we klaar om het te analyseren.
wat nu?
u kunt nu de opgehaalde gegevens analyseren met behulp van alle sequentieanalysetools en workflows in Galaxy. SRA houdt steungegevens voor elk denkbaar type van *-seq experiment.
Als u deze tutorial hebt uitgevoerd, maar datasets hebt opgehaald waarin u geïnteresseerd was, raadpleeg dan de rest van de GTN-bibliotheek voor ideeën over het analyseren in Galaxy.
echter, als u de datasets hebt opgehaald die in bovenstaande voorbeelden van deze tutorial zijn gebruikt, dan bent u klaar om de SARS-CoV-2 variantanalyse hieronder uit te voeren.
Variatieanalyse van SARS-Cov-2 sequentiegegevens
In dit deel van de tutorial zullen we variantaanroepen en basisanalyses uitvoeren van de hierboven gedownloade datasets. We zullen beginnen met het downloaden van de Wuhan-Hu-1 SARS-CoV-2 referentie sequentie, vervolgens uitvoeren adapter trimmen, uitlijning en variant bellen en ten slotte kijken naar de geografische verdeling van een aantal van de gevonden varianten.
commentaar op de usegalaxy.* Covid-19 analyse project
Deze tutorial gebruikt een subset van de gegevens en loopt door de variatieanalysesectie van covid19.galaxyproject.organisatie.De gegevens voor covid19.galaxyproject.org wordt voortdurend bijgewerkt naarmate nieuwe datasets openbaar worden gemaakt.
haal de referentie-genoomgegevens
de referentie-genoomgegevens voor vandaag zijn voor SARS-CoV-2, “Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome”, met de toetredingsidentificatie van NC_045512.2.
deze gegevens zijn beschikbaar via Zenodo via de volgende link.
hands_on Hands-on: Get the reference genome data
importeer het volgende bestand in uw geschiedenis:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzTip: Importeren via links
- Kopieer de locatie van de link
- Open de Galaxy Upload Manager (galaxy-upload op de rechterbovenhoek van het instrument paneel)
- Kies Plakken/Ophalen van Gegevens
- Plak de link in het tekstveld
- Druk op Start
- Sluit de windowBy standaard, Galaxy maakt gebruik van de URL als de naam, zodat de naam van de bestanden met een naam.
adapter trimmen met fastp
verwijderen van sequencing adapters verbetert uitlijningen en variantenaanroepen. het fastp-hulpmiddel kan wijd gebruikte het rangschikken adapters automatisch ontdekken.
hands_on Hands-on: Task description
- fastp tool met de volgende parameters:
- “Single-end or paired reads”:
Paired Collection
- param-file”select paired collection(s)”:
list_paired(uitgang van de Snellere Download en Extract Leest in FASTQ tool)- In “uitvoeropties”:
- Output “JSON-rapport”:
Yes
Uitlijning met de Kaart met de BWA-MEM
BWA-MEM-tool is een veel gebruikte volgorde aligner voor korte-lees sequencing datasets zoals die we analyseren in deze tutorial.
hands_on Hands-on: Sequencing reads uitlijnen naar referentie-genoom
- kaart met BWA-MEM tool met de volgende parameters:
- “zal u een referentie-genoom uit uw geschiedenis selecteren of een ingebouwde index gebruiken?”:
Use a genome from history and build index
- param-file” Use the following dataset as the reference sequence”:
output(Input dataset)- ” Single or Paired-end reads”:
Paired Collection
- param-file” select a paired collection”:
output_paired_coll(output van Fastp tool)- ” set Read groups information?”:
Do not set- “select analysis mode”:
1.Simple Illumina mode
duplicaten verwijderen met Markduplicaten
Markduplicaten tool verwijdert dubbele sequenties afkomstig van bibliotheekvoorbereidingsartefacten en artefacten rangschikken. Het is belangrijk om deze artefactual opeenvolgingen te verwijderen om kunstmatige oververtegenwoordiging van enige molecule te vermijden.
hands_on Hands-on: Verwijder PCR duplicaten
- MarkDuplicates tool met de volgende parameters:
- param-file “select SAM/BAM dataset or dataset collection”:
bam_output(output of Map with BWA-MEM tool)- “If true schrijf geen duplicaten naar het uitvoerbestand in plaats van ze met de juiste vlaggen te schrijven”:
Yes
genereer uitlijningsstatistieken met samtools stats
na de dubbele markering hierboven kunnen we statistieken genereren over de uitlijning die we hebben gegenereerd.
hands_on Hands-on: Genereren uitlijning statistieken
- Samtools stats tool met de volgende parameters:
- param-bestand “BAM bestand”:
outFile(uitgang van de MarkDuplicates tool)- “Set dekking distributie”:
No- “Output”:
One single summary file- “Filter door SAM vlaggen”:
Do not filter- “Gebruik een referentie sequentie”:
No- “Filteren op regio ‘s”:
No
Realign reads met lofreq viterbi
Realign reads tool corrigeert misalignments rond inserties en deleties. Dit is nodig om varianten nauwkeurig te kunnen detecteren.
hands_on Hands-on: Realign reads around indels
- Realign reads with lofreq tool with the following parameters:
- param-file “Reads to realign”:
outFile(output of MarkDuplicates tool)- “kies de bron voor het referentie genoom”:
History
- param-file “Reference”:
output(Input dataset)- In” Advanced options”:
- ” Hoe omgaan met basiskwaliteiten van 2?”:
Keep unchanged
voeg indel-kwaliteiten toe met lofreq voeg indel-kwaliteiten
Deze stap voegt indel-kwaliteiten toe aan ons uitlijningsbestand. Dit is nodig om varianten aan te roepen met behulp van Aanroepvarianten met lofreq tool
hands_on Hands-on: Toevoegen indel kwaliteiten
- Invoegen indel kwaliteiten met lofreq tool met de volgende parameters:
- param-bestand “Leest”:
realigned(de output van het Uitlijnen van de leest tool)- “Indel berekening aanpak”:
Dindel
- “Kies de bron voor de referentie genoom”:
History
- param-bestand “Reference”:
output(Input dataset)
Oproep Varianten met lofreq Oproep varianten
We zijn nu klaar om te bellen varianten.
hands_on Hands-on: Bel varianten
- Oproep varianten met lofreq tool met de volgende parameters:
- param-bestand “Input leest in het BAM-indeling”:
output(uitgang van de Plaats indel kwaliteiten tool)- “Kies de bron voor de referentie genoom”:
History
- param-bestand “Reference”:
output(Input dataset)- “Call varianten over”:
Whole reference- Soorten varianten te noemen”:
SNVs and indels- “Variant bellen parameters”:
Configure settings
- In “Dekking” voor:
- “Minimale dekking”:
50- In “Base-bellen”:
- Minimum baseq”:
30- Minimum baseq voor alternatieve bases”:
30- in het ‘mapping qualityy
20- “variant filter parameters”:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
De uitvoer van deze stap is een verzameling VCF-bestanden die kunnen worden gevisualiseerd in een genoombrowser.
annoteer variante effecten met SnpEff eff:
We zullen nu de varianten die we in de vorige stap noemden annoteren met het effect dat ze hebben op het SARS-CoV-2 genoom.
hands_on Hands-on: annoteer varianteffecten
- SnpEff eff: gereedschap met de volgende parameters:
- param-bestand “Sequence changes (SNPs, MNPs, InDels)”:
variants(uitgang van de Oproep varianten tool)- “Output format”:
VCF (only if input is VCF)- “Maak CSV-rapport, bruikbaar voor verdere analyse (-csvStats)”:
Yes- “Annotatie opties”: `
- “Filter output”: `
- “Filter specifieke Effecten”:
No
De uitvoer van deze stap is een VCF-bestand met toegevoegde variant effecten.
Creëer een tabel met varianten met behulp van Snpsift-Extractvelden
We zullen nu verschillende effecten uit de VCF selecteren en een tabelbestand maken dat gemakkelijker te begrijpen is voor mensen.
hands_on Hands-on: Create table of variants
- SnpSift Extract Fields tool with the following parameters:
- param-file “Variant input file in VCF format”:
snpeff_output(output of SnpEff EVF: tool)- “Fields uitpakken van”:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- “multiple field separator”:
,- “lege veld tekst”:
.
We kunnen inspecteren van de output-bestanden en zie controleren of Varianten in dit bestand worden ook beschreven in een waarneembare notebook dat toont de geografische verdeling van de SARS-CoV-variant 2 reeksen
Interessante varianten zijn de C-T variant op positie 14408 (14408C/T) in SRR11772204, 28144T/C in SRR11597145 en 25563G/T in SRR11667145.
vat gegevens samen met MultiQC
We zullen nu onze Analyse samenvatten met MultiQC, wat een mooi rapport voor onze gegevens genereert.
hands_on Hands-on: Resumize data
- MultiQC tool with the following parameters:
- In “Results”:
- param-repeat “Insert Results”
- ” welk tool werd gebruikt genereer logs?”:
fastp
- param-file “Output of fastp”:
report_json(uitvoer van fastp tool)- param-herhaal “Insert Results”
- “welk gereedschap werd gebruikt genereer logs?”:
Samtools
- In “Samtools output”:
- param-repeat “Insert Samtools output”
- ” Type Samtools output?”:
stats
- param-file “Samtools stats output”:
output(output van samtools stats tool)- param-herhaal “Insert Results”
- ” welk gereedschap werd gebruikt genereer logs?”:
Picard
- In “Picard output”:
- param-repeat” Insert Picard output “
- ” Type Picard output?”:
Markdups- param-file “Picard output”:
metrics_file(output van het gereedschap Markduplicaten)- param-herhaal “resultaten invoegen”
- ” welk gereedschap werd gebruikt om logs te genereren?”:
SnpEff
- param-bestand “- Uitgang van SnpEff”:
csvFile(uitgang van de SnpEff eff: tool)
Conclusie
Gefeliciteerd, je weet nu hoe het importeren van sequencedata van de SRA en het uitvoeren van een voorbeeld van een analyse van deze datasets.
toetspunten sleutelpunten
Sequentiegegevens in de SRA kunnen direct worden geïmporteerd in Galaxy
Veelgestelde vragen
heeft u vragen over deze tutorial? Kijk op de FAQ pagina voor de variant analyse onderwerp om te zien of uw vraag staat daar. Zo niet, stel dan uw vraag op het GTN Gitter kanaal of het Galaxy Help Forum
nuttige literatuur
meer informatie, inclusief links naar documentatie en originele publicaties, met betrekking tot de tools, analysetechnieken en de interpretatie van de resultaten die in deze tutorial worden beschreven, kunt u hier vinden.
Feedback
heeft u dit materiaal gebruikt als instructeur? Voel je vrij om ons feedback te geven over hoe het ging.
Citing this Tutorial
- Marius Van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 From NCBI ‘ s Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials). / training-materiaal / onderwerpen / variant-analyse/tutorials/sars-cov-2 / tutorial.html Online; vandaag geopend
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology cels Systems 10.1016/j.cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}