aminokwasy, nukleotydy lub jakikolwiek inny charakter ewolucyjny są w pewnym stopniu zastępowane przez inne. Na przykład wyobraźmy sobie sekwencję ewolucyjną z trzema możliwymi Stanami, A, B i C. Jeśli model podstawienia jest odwracalny w czasie, będą trzy szybkości przejścia, a<>B, B<>C i a<> C.
Załóżmy, że stawki wynoszą odpowiednio 1, 1 i 0 w jednostkach podstawienia na 100 znaków na jednostkę czasu. Po jednej jednostce czasu, w 300 znakowej długiej sekwencji pierwotnie składającej się w równym stopniu z As, Bs i Cs, spodziewamy się, że będzie jedno podstawienie A do B i jedno podstawienie B do C. Jeśli porównujemy dwie sekwencje homologiczne w organizmach żywych, ponieważ jedna jednostka czasu przypadła na obie sekwencje, spodziewamy się dwóch sekwencji a do B i dwóch B do Csubstytucji pomiędzy dzisiejszymi sekwencjami.
bez względu na to, jak długo prowadzimy ten proces, nigdy nie będzie bezpośredniego zastąpienia A przez C. nigdy nie będzie również podstawienia od A do C w ASO-zwanym modelem nieskończonych miejsc, w którym w jednym miejscu może wystąpić nie więcej niż jedno podstawienie.
ponieważ jednak podstawienia A do B i B do C są powszechne, w skończonych miejscach model ostatecznie B zostanie zastąpiony przez C w miejscu, w którym A było wcześniej umieszczone przez B. To pośrednie zastąpienie a przez C (lub równoważnie w modelu odwracalnym czasowo, C przez a) staje się bardziej prawdopodobne, im dłuższy okres czasu oddziela sekwencje homologiczne.
symulowałem ewolucję sekwencji w oparciu o powyższy scenariusz, uruchamiając symulację przez 10 jednostek czasu. Z tego podstawienia zaobserwowałem następujące liczby dla każdego wzorca miejsca:
| A | B | C | |
|---|---|---|---|
| A | 91 | 9 | 0 |
| B | 5 | 86 | 9 |
| C | 0 | 9 | 91 |
w tym relatywnie krótkim czasie nie wydaje się, aby wystąpił jakiś<>csubstytucje. Jednak kiedy powtórzę symulację na 100 jednostek czasu:
| A | B | C | |
|---|---|---|---|
| A | 55 | 35 | 10 |
| B | 29 | 36 | 35 |
| C | 20 | 36 | 44 |
jak widać, wiele znaków „a” zostało zastąpionych przez „C” i viceversa. Bardziej ogólnie, w modelu skończonych miejsc wielokrotne podstawienia, ponieważ rozkład liczby wzorców miejsc staje się znacznie bardziej płaski poza tym, co znacznie zwiększa udział liczby poza przekątnej w stosunku do liczby przekątnej.Macierze punktowe Pam i BLOSUM odpowiadają za wiele podstawień w różny sposób.
matryce PAM dla aminokwasów, wraz z jednoliterowymi skrótami używanymi dla genetycznie kodowanych aminokwasów, zostały opracowane przez MargaretDayhoff. Zostały one pierwotnie opublikowane w 1978 roku, a na podstawie proteinsequences Dayhoff kompilował od 1960 roku, opublikowane jako theAtlas of Protein Sequence and Structure.
nazwa PAM pochodzi od „point accepted mutation” i odnosi się do połączenia jednego aminokwasu w białku z innym aminokwasem.Mutacje te zidentyfikowano przez porównanie bardzo podobnych sekwencji z co najmniej 85% tożsamością i zakłada się, że wszelkie zaobserwowane substytucje były wynikiem pojedynczej mutacji między sekwencją przodków a jedną z obecnych sekwencji dnia.
Pam definiuje również jednostkę czasu, gdzie 1 PAM to czas, w którym 1/100 aminokwasów może ulec mutacji. Macierz prawdopodobieństwa PAM1 pokazuje prawdopodobieństwo, że aminokwas w kolumnie j zostanie zastąpiony aminokwasem w wierszu I. obliczono go na podstawie liczby Pam Dayhoffa i przeskalowano na 1 jednostkę Pam czasu. Jak widać, prawdopodobieństwa poza przekątną macierzy pam1 są bardzo małe (wszystkie elementy zostały skalowane przez 10 000 dla możliwościlegalności):
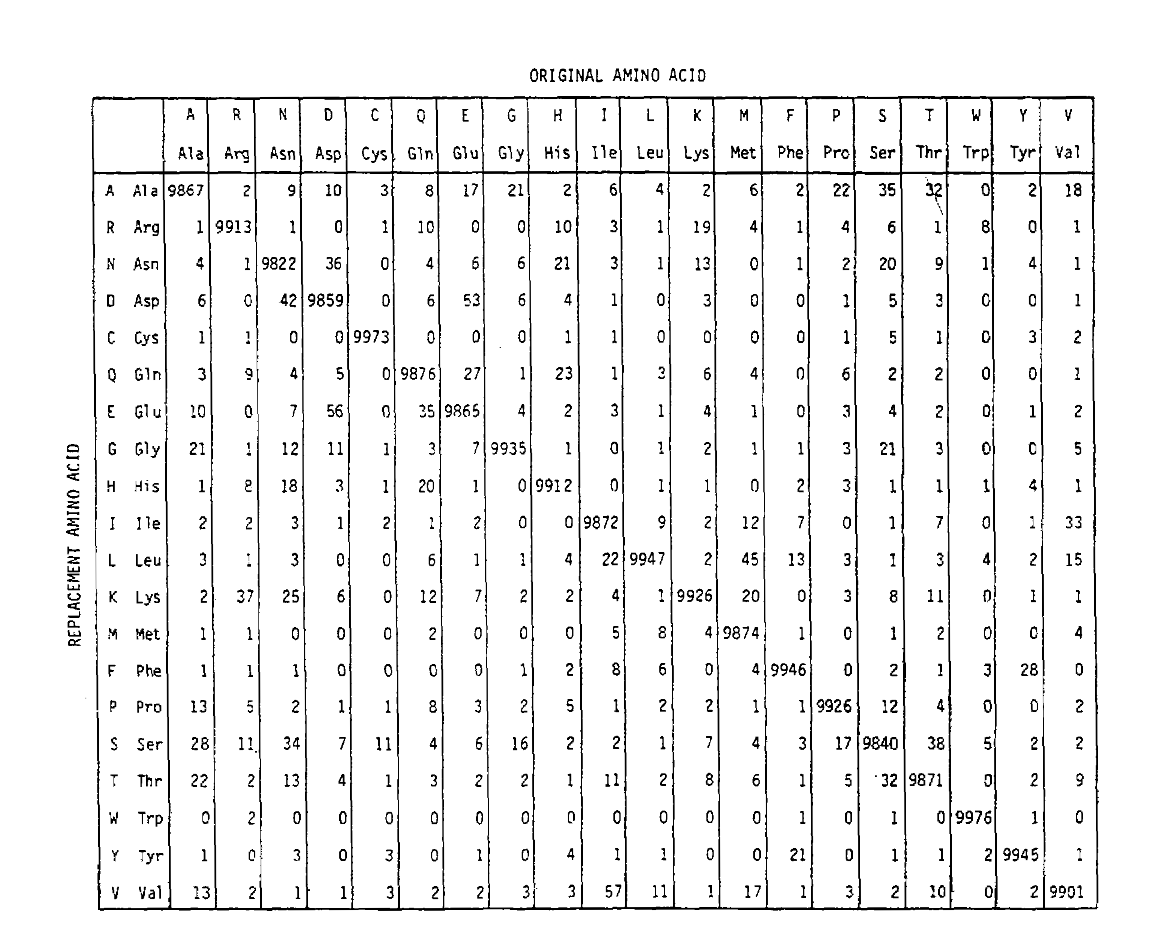
aby obliczyć prawdopodobieństwo wymiany aminokwasu dla dłuższych czasów, macierz może być pomnożona przez samą siebie przez odpowiadającą jej liczbę razy. W ten sposób macierz prawdopodobieństwa PAM250, opisująca prawdopodobieństwo przesunięcia danych o 250 jednostkach PAM czasu, została wyprowadzona przez podniesienie macierzy prawdopodobieństwa PAM1 do potęgi 250 (wszystkie elementy zostały skalowane przez 100 dla czytelności):
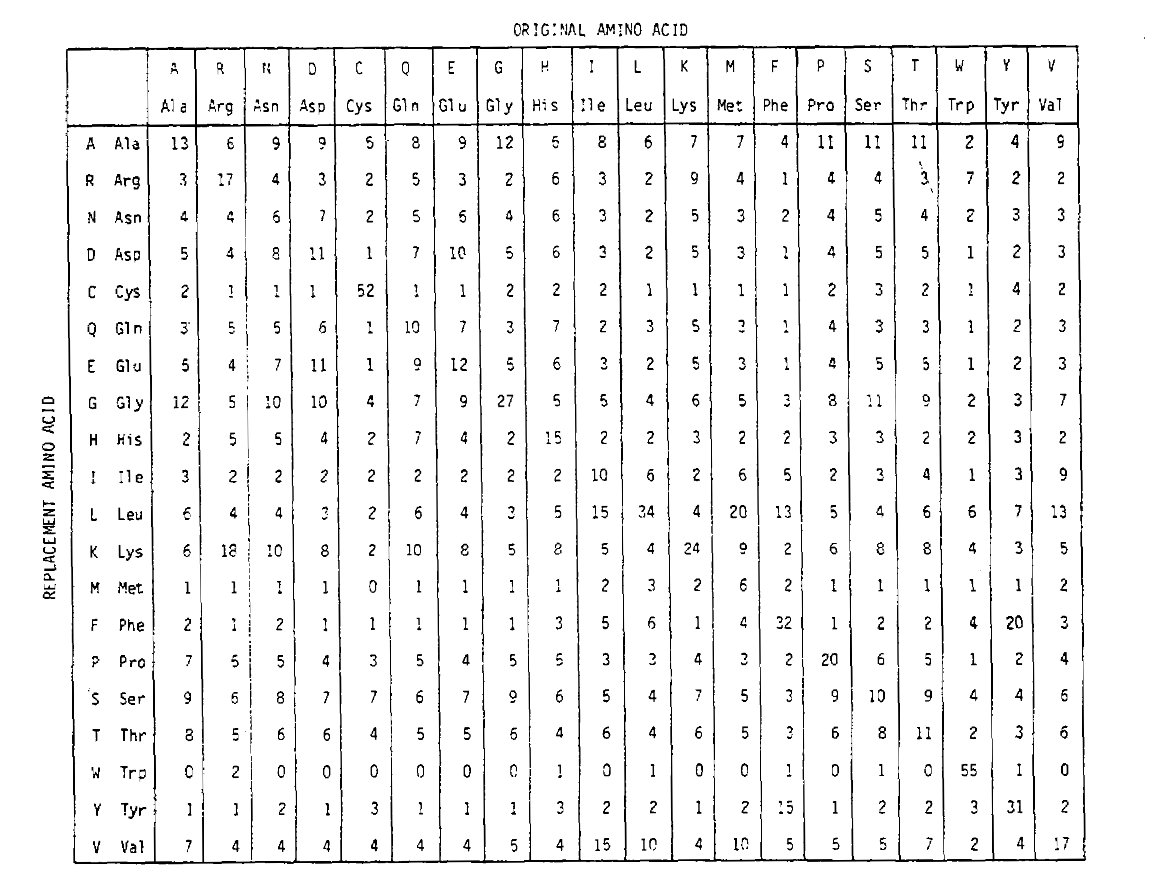
Nie tylko są one proporcjonalnie większe, jak można by się spodziewać przez dłuższy czas, ale są bardziej płaskie. Na przykład prawdopodobieństwo wymiany waliny (V)na izoleucynę (i) jest 33× większe niż wymiany V na histadynę (H)w macierzy PAM1, ale tylko 4,5× większe w macierzy PAM250.
macierze punktowe można następnie obliczyć z macierzy prawdopodobieństwa i częstotliwości bazowych.
matryce BLOSUM, opracowane przez Stevena i Jorję Henikoffów i opublikowane w 1992 roku, mają zupełnie inne podejście. Podczas gdy Pam jest domyślnie stosowany w stacjonarnym modelu ewolucji z wykorzystaniem wykładników macierzowych, efekt wielokrotnych podstawień jest rozpatrywany pośrednio w BLOSUM przez konstruowanie różnych macierzy punktowych dla różnych skal czasowych.
w obrębie wielu sekwencji sekwencji homologicznych identyfikuje się zachowane bloki aminokwasów. W obrębie każdego bloku multipleksy są grupowane, gdy ich parami średnia tożsamość sekwencji jest wyższa niż jakiś próg. Próg wynosi 80% dla matrycy BLOSUM80, 62% dla BLOSUM62, 50% dla BLOSUM50 i tak dalej.
oznacza to, że dla BLOSUM80 bloki będą miały średnią identyczność par nie większą niż 80%, dla BLOSUM62 nie większą niż 62%, itd.
prawdopodobieństwo wymiany aminokwasów dla sekwencji homologicznych oblicza się na podstawie porównań par pomiędzy klastrami. Prawdopodobieństwo to będzie wynikało z pojedynczych i wielokrotnych podstawień, przy czym wielokrotne podstawienia będą miały większy wpływ na większe odległości ewolucyjne. Dlatego też wskaźniki wyników generowane na podstawie porównań par pomiędzy klastrami o średniej odległości większej, jak macierz BLOSUM50, będą w naturalny sposób odpowiadać za większy efekt wielokrotnych podstawień.
mimo, że obierają różne trasy, matura końcowa BLOSUM i Pam score są właściwie całkiem podobne. Według Henikoffa i Henikoffa następujące macierze Pam i BLOSUM są porównywalne:
| PAM | BLOSUM |
|---|---|
| PAM250 | BLOSUM45 |
| PAM160 | BLOSUM62 |
| PAM120 | BLOSUM80 |
For more information on PAM (Dayhoff) and BLOSUM matrices, see Rozdział 2 biologiczna analiza sekwencji przez Durbin et al., oraz Wikipedia.
aktualizacja 13 października 2019: aby zapoznać się z inną perspektywą macierzy zastępczych, zapoznaj się z sekcją „objazdy” na końcu rozdziału 5 algorytmów bioinformatycznych (wydanie 2 lub 3) autorstwa Compeau i Pevznera.