celem tego poradnika jest wprowadzenie do przetwarzania danych sekwencjonowania nowej generacji w galaktyce. Ten samouczek używa wywołania wariantu COVID-19 z danych Illumina, ale nie chodzi o wywołanie wariantu jako takiego.
Po zakończeniu tego kursu dowiesz się:
- Jak znaleźć dane w SRA i przesłać te informacje do Galaxy
- Jak wykonać podstawowe przetwarzanie danych NGS W Galaxy, w tym:
- Kontrola jakości (QC) danych Illumina
- mapowanie
- usuwanie duplikatów
- wywołanie wariantu z
lofreq - adnotacja wariantu
- korzystanie ze zbiorów zbiorów danych
- Importowanie danych do Jupyter
### Agenda>> w tym samouczku omówimy:>> 1. TOC> {:toc}> {: .agenda} # # dwie ścieżki w tym tutorialu stworzyliśmy dwie trajektorie, które możesz śledzić w tym tutorialu.1. ** Trajektoria 1 * * – Zacznij od SRA NCBI i Wyszukaj dostępne akcesoria → Start (#the-sequence-read-archive)2. ** Trajektoria 2* * – Omiń SRA NCBI i zacznij bezpośrednio od galaktyki. → Start (#back-in-galaxy)zalecamy rozpoczęcie od * * trajektorii 2**.# Sekwencja odczytu Archiwum (https://www.ncbi.nlm.nih.gov/sra) jest podstawowym archiwum *niezmontowanych odczytów* dla (https://www.ncbi.nlm.nih.gov/). SRA jest doskonałym miejscem, aby uzyskać dane sekwencjonowania, które leżą u podstaw publikacji i badań.Ten samouczek opisuje, jak uzyskać dane sekwencyjne z SRA do galaktyki za pomocą bezpośredniego połączenia między nimi.> ### comment Skomentuj>> usłyszysz również SRA o nazwie *Short Read Archive*, jego oryginalną nazwę.> {:.comment}# # dostęp do SRASRY można uzyskać bezpośrednio przez jego stronę internetową lub za pośrednictwem panelu narzędzi na Galaxy.> ### comment Komentarz>> początkowo opcja panelu narzędzi do dostępu do SRA istnieje tylko na (https://usegalaxy.org/). Wsparcie dla bezpośredniego połączenia z SRA będzie zawarte w wydaniu 20.05 Galaxy{: .komentarz}> ### hands_on Hands-on: Explore SRA Entrez>> 1. Przejdź do wybranej instancji Galaxy, takiej jak jedna z (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) lub dowolna inna. (Ten poradnik wykorzystuje usegalaxy.org).> 1. Jeśli Twoja historia nie jest jeszcze pusta, rozpocznij nową historię (zobacz (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html), aby uzyskać więcej informacji na temat historii galaktyk)> 1. ** Kliknij * * „Pobierz dane” w górnej części panelu Narzędzia.> 1. ** Kliknij * * „Sra Server” na liście narzędzi wyświetlanych w sekcji „Pobierz dane”.> to zabierze cię do (https://www.ncbi.nlm.nih.gov/sra) — możesz również uruchomić bezpośrednio z SRA. Pole wyszukiwania jest wyświetlane na górze strony. Spróbuj wyszukać coś, co Cię interesuje, na przykład „delfin” lub „nerka” lub „nerka delfina”, a następnie **kliknij** przycisk „Szukaj”.>> zwraca listę eksperymentów *SRA*, które pasują do szukanego ciągu. Eksperymenty SRA, znane również jako * wpisy SRX*, zawierają dane sekwencyjne z konkretnego eksperymentu, a także wyjaśnienie samego eksperymentu i wszelkich innych powiązanych danych. Możesz zbadać zwrócone eksperymenty, klikając ich nazwę. Zobacz (https://www.ncbi.nlm.nih.gov/książki/NBK56913/) w (https://www.ncbi.nlm.nih.gov/książki/N/helpsrakb/), aby uzyskać więcej.>> wprowadzając tekst w polu wyszukiwania SRA, używasz (https://www.ncbi.nlm.nih.gov/sra/docs/srasearch/). Entrez obsługuje zarówno proste wyszukiwania tekstowe, jak i bardzo precyzyjne wyszukiwania, które sprawdzają określone metadane i używają arbitralnie złożonych wyrażeń logicznych. Entrez pozwala na skalowanie Wyszukiwania od podstawowego do zaawansowanego, jak zawęzić wyszukiwania. Składnia zaawansowanych wyszukiwań może wydawać się zniechęcająca, ale SRA zapewnia graficzną (https://www.ncbi.nlm.nih.gov/sra/advanced/) do wygenerowania określonej składni. I, jak zobaczymy poniżej, Sra Run Selector zapewnia jeszcze bardziej przyjazny interfejs użytkownika do zawężania wybranych danych.>> baw się interfejsem Sra Entrez, w tym zaawansowanym konstruktorem zapytań, aby sprawdzić, czy możesz zidentyfikować zestaw eksperymentów SRA, które są istotne dla jednego z obszarów badawczych.{: .hands_on}> # # # hands_on Hands-on: Wygeneruj listę pasujących eksperymentów za pomocą Entrez>> teraz, gdy masz podstawową znajomość Sra Entrez, znajdźmy sekwencje używane w tym samouczku.>> 1. Jeśli jeszcze tam nie jesteś, **przejdź** z powrotem do (https://www.ncbi.nlm.nih.gov/sra> 1. ** Wyczyść * * dowolny tekst wyszukiwania z pola wyszukiwania.> 1. ** Wpisz * * „sars-cov-2” w polu wyszukiwania i **kliknij** „Szukaj”.> to zwraca długą listę eksperymentów SRA, które pasują do naszego Wyszukiwania, a ta lista jest zbyt długa, aby użyć jej w ćwiczeniu samouczka. W tym momencie możemy użyć zaawansowanego konstruktora zapytań Entrez, o którym dowiedzieliśmy się powyżej.> ale nie będziemy. zamiast tego wysyłamy listę wyników * too long for a tutorial* do selektora Sra Run i używamy jego przyjaznego interfejsu, aby zawęzić nasze wyniki.>> !(../../ images / sra_entrez.png) {:.hands_on}> # # # hands_on Hands-on: Przejdź z Entrez do Sra Run Selector>> Wyświetl wyniki jako rozszerzoną tabelę interaktywną za pomocą Runselectora.>> 1. Kliknij Wyślij wyniki, aby uruchomić selektor, który pojawi się w polu u góry wyników wyszukiwania.>> !(../../ images / sra_entrez_result.png)>>> ### wskazówka co zrobić, jeśli nie widzisz linku wyboru uruchamiania?>>>> być może zauważyłeś ten tekst wcześniej, gdy przeglądałeś Wyszukiwanie Entrez. Ten tekst pojawia się tylko w niektórych przypadkach, gdy liczba wyników wyszukiwania mieści się w dość szerokim oknie. Nie zobaczysz go, jeśli masz tylko kilka wyników, i nie zobaczysz go, jeśli masz więcej wyników niż Selektor biegu może zaakceptować.>>>> *aby wysłać wyniki Do Galaxy, musisz uruchomić Selektor.* Co zrobić, jeśli nie masz wystarczająco dużo wyników, aby uruchomić ten link jest wyświetlany? W takim przypadku wywołujesz get to the Run Selector poprzez* * kliknięcie * * w menu rozwijanym „Wyślij do” w prawym górnym rogu panelu wyników. Aby przejść do wyboru biegu, * * wybierz * * „wybór biegu”, a następnie **kliknij * * przycisk „Przejdź”.> !(../../ images / sra_entrez_send_to.png)> {: .tip}>>> 1. ** Kliknij * * 'Wyślij wyniki do uruchomienia selektora’ w górnej części panelu wyników wyszukiwania. (Jeśli nie widzisz tego linku, zobacz komentarz bezpośrednio powyżej.){: .hands_on}## Sra Run Selectorjednak nauczyliśmy się wcześniej, jak zawęzić wyniki wyszukiwania przy użyciu zaawansowanej składni Entrez. Jednak nie skorzystaliśmy z tej mocy, gdy byliśmy w Entrez. Zamiast tego użyliśmy prostego wyszukiwania, a następnie wysłaliśmy wszystkie wyniki do selektora biegu. Nie mamy jeszcze (krótkiej) listy wyników, na których chcemy przeprowadzić analizę. * What are we doing?* Używamy Entrez I Run Selector, jak są one przeznaczone do użycia: * Użyj interfejsu Entrez, aby zawęzić wyniki do rozmiaru, który Selektor Run może zużywać. * Wyślij te wyniki Entrez do selektora Sra Run * użyj bardziej przyjaznego interfejsu selektora Run do 1. Łatwiej zrozumieć dane, które mamy 1. Zawęzić te wyniki za pomocą tej wiedzy.> ### comment Run Selector jest zarówno większy, jak i mniejszy niż Entrez>> Run Selector może zrobić większość, ale nie wszystko, co może zrobić składnia wyszukiwania Entrez. Run selector wykorzystuje technologię* faceted search*, która jest łatwa w użyciu i wydajna, ale ma wrodzone ograniczenia. W szczególności, Entrez będzie działać lepiej podczas wyszukiwania atrybutów, które mają dziesiątki, setki lub tysiące różnych wartości. Uruchom Selektor będzie działać lepiej atrybuty wyszukiwania z mniej niż 20 różnych wartości. Na szczęście opisuje to większość poszukiwań.{: .komentarz}okno wyboru biegu jest podzielone na kilka paneli:* **`Lista filtrów`**: w lewym górnym rogu. To tutaj udoskonalimy nasze poszukiwania.** * ` Select’**: podsumowanie tego, co zostało początkowo przekazane do run Selector i ile z tego wybraliśmy do tej pory. (I do tej pory nie wybraliśmy żadnego z nich.) Zwróć również uwagę na kuszący, ale nadal wyszarzony przycisk „Galaxy”.** * ` Znaleziono Elementy x ’ * * początkowo jest to lista elementów wysłanych do uruchomienia selektora z Entrez. Ta lista będzie się kurczyć, gdy zastosujemy do niej filtry.!(../../ images / sra_run_selector.png)> ### komentarz dlaczego liczba znalezionych pozycji *wzrosła?* >> Przypomnijmy, że interfejs Entrez zawiera listę eksperymentów SRA (wpisy SRX). Lista Run selector * runs — – sekwencjonowanie zestawów danych-i istnieje * jeden lub więcej * runs na eksperyment. Mamy te same dane, co wcześniej, teraz widzimy je w najdrobniejszych szczegółach.{: .comment} `Lista filtrów ’ w lewym górnym rogu pokazuje kolumny w naszych wynikach, które mają albo ciągłe wartości liczbowe, albo 10 lub mniej (możesz zmienić tę liczbę) różne wartości w nich. ** Przewiń * * w dół listy wybierz kilka filtrów. Po wybraniu filtra, poniżej pojawi się pole* wartości*, lista opcji tego filtra i liczba uruchomień z każdą opcją. Te wartości / opcje są pobierane z metadanych zestawu danych. Spróbuj * * wybierając * * kilka ciekawie brzmiących filtrów, a następnie * * wybierz * * jedną lub więcej opcji dla każdego filtra. Spróbuj * * odznaczyć * * opcje i filtry. Jak to zrobić, liczba znalezionych wyników zmniejszy się lub wzrośnie.> ### tip Tip: Użyj filtrów, aby lepiej zrozumieć dane>> filtry są sposobem zawężania zbiorów danych rozważanych do wysłania do Galaxy, ale są również doskonałym sposobem na zrozumienie danych:> po pierwsze, wybór filtra jest łatwym sposobem na sprawdzenie zakresu wartości w kolumnie. Możesz nie być w stanie (https://www.google.com/search?q=sra+sirs_outcome), ale możesz dowiedzieć się, widząc, jakie wartości są w nim.> po drugie, możesz zbadać, jak różne kolumny odnoszą się do siebie. Czy istnieje związek pomiędzy wartościami „sirs_outcome” a wartościami „disease_stage”?{: .tip} > ### hands_on Hands-on: Zawęź wyniki za pomocą selektora Run>> 1. Jeśli masz włączone filtry,** odznacz je**.> gdy to zrobisz, pod `listą filtrów`Nie pojawią się Żadne pola *values*.> 2. ** Skopiuj i wklej * * ten ciąg wyszukiwania w polu wyszukiwania „znalezione przedmioty”.>> SRR11772204 lub SRR11597145 lub SRR11667145>> ta ręka-wybrany zestaw biegów ogranicza nasze wyniki do 3 biegów z różnych dystrybucji geograficznych.{: .hands_on} zmniejsza to listę znalezionych przedmiotów z dziesiątek tysięcy biegów do 3 biegów (łatwa do opanowania Liczba dla samouczka!). Ale nie skończyliśmy jeszcze z Run Selector. Pamiętaj, że Przycisk „Galaktyka” jest nadal wyszarzony. Zawęziliśmy nasze opcje, ale nie wybraliśmy jeszcze niczego do wysłania do galaktyki.Możliwe jest wybranie każdego pozostałego biegu, klikając *** znacznik wyboru u góry pierwszej kolumny. Możesz odznaczyć wszystko, klikając ` * * * „X”.> ### hands_on Hands-on: wybierz Uruchom i wyślij do galaktyki>> 1. Wybierz wszystkie biegi, klikając ` * * * „X”.> I Teraz przycisk `Galaxy` jest aktywny.> 1. ** Kliknij * * przycisk „Galaxy” w sekcji „Wybierz” u góry strony.{: .hands_on}# # powrót do Galaktykgdy klikamy 'Galaxy’ w selektorze Run dzieje się kilka rzeczy. Po pierwsze, uruchamia nową kartę przeglądarki lub okno, które otwiera się w Galaxy. Zobaczysz * duże zielone pole * wskazujące, że uścisk dłoni między SRA A Galaxy powiódł się, a następnie zobaczysz nowe zadanie ” SRA ” w panelu historii. To pole może zaczynać się jako szary / oczekujący, wskazując, że transfer jeszcze się nie rozpoczął, lub może przejść od razu do żółtego / running lub Zielonego / done.> ### hands_on Hands-on: Sprawdź nowy zestaw danych SRA>> 1. Po zakończeniu transferu ” SRA ” ** kliknij * * na ikonę galaxy-eye (oko) zestawu danych.>> to wyświetla zestaw danych w centralnym panelu galaktyki.{: .hands_on}zestaw danych ’ SRA ’ nie jest danymi sekwencyjnymi, ale raczej *metadanymi*, których użyjemy, aby uzyskać dane sekwencyjne z SRA. Te metadane odzwierciedlają informacje, które widzieliśmy w sekcji „znalezione elementy” selektora biegu. Metadane nie są danymi końcowymi, których szukamy w SRA, ale posiadanie wszystkich tych metadanych jest często przydatne w kolejnych etapach analizy.Użyjmy tych metadanych do pobrania danych sekwencji z SRA. SRA zapewnia narzędzia do wydobywania wszelkiego rodzaju informacji, w tym samych danych sekwencyjnych. Narzędzie Galaxy 'Faster Download and Extract Reads in FASTQ’ jest oparte na narzędziu Sra (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump) I właśnie to robi.– >
- Znajdź niezbędne dane w SRA
- hands_on Hands-on: opis zadania
- skomentuj komentarz
- proces i filtr SraRunInfo.plik csv w Galaxy
- hands_on Hands-on: Upload SraRunInfo.plik csv do galaktyki
- komentarz uwaga na cięcia
- hands_on Hands-on: Tworzenie podzbioru danych
- wskazówka: Znajdź narzędzia
- Pobierz dane sekwencjonowania z szybszym pobieraniem i wyciąganiem w FASTQ
- hands_on Hands-on: opis zadania
- co teraz?
- Analiza zmienności danych sekwencjonowania SARS-Cov-2
- skomentuj* Projekt analizy COVID-19
- Pobierz referencyjne dane genomu
- hands_on Hands-on: Get the reference genome data
- Wskazówka: Importowanie za pomocą linków
- przycinanie adapterów za pomocą Fastp
- hands_on Hands-On: opis zadania
- wyrównanie z mapą za pomocą BWA-mem
- hands_on Hands-on: Wyrównaj odczyty sekwencjonowania do genomu referencyjnego
- Usuń duplikaty za pomocą MarkDuplicates
- hands_on Hands-on: Usuń duplikaty PCR
- Generuj statystyki wyrównania za pomocą samtools stats
- hands_on Hands-on: Generowanie statystyk wyrównania
- Realign odczytuje z lofreq viterbi
- hands_on Hands-on: Realign odczytuje indele
- Dodaj jakość indel za pomocą lofreq Wstaw jakość indel
- hands_on Hands-on: Dodaj jakość indel
- warianty wywołania korzystając z opcji wywołania lofreq
- hands_on Hands-on: warianty połączeń
- opisywanie efektów wariantowych za pomocą SnpEff eff:
- hands_on Hands-on: Adnotuj efekty wariantu
- Tworzenie tabeli wariantów za pomocą SnpSift Extract Fields
- hands_on Hands-on: Utwórz tabelę wariantów
- podsumowanie danych za pomocą MultiQC
- hands_on Hands-on: podsumowanie danych
- wnioski
- punkty kluczowe punkty
- Często zadawane pytania
- przydatna Literatura
- Feedback
- powołując się na ten poradnik
- details BibTeX
Znajdź niezbędne dane w SRA
najpierw musimy znaleźć dobry zestaw danych do zabawy. Archiwum odczytu sekwencji (ang. Sequence Read Archive, Sra) – podstawowe archiwum niezmontowanych odczytów, zarządzane przez amerykańskie Narodowe Instytuty Zdrowia (NIH). SRA jest doskonałym miejscem, aby uzyskać dane sekwencjonowania, które leżą u podstaw publikacji i badań. Zróbmy to:
hands_on Hands-on: opis zadania
- przejdź do strony SRA NCBI, wskazując przeglądarkę nahttps://www.ncbi.nlm.nih.gov/sra
- w polu wyszukiwania wpisz
SARS-CoV-2 Patient Sequencing From Partners / MGH(alternatywnie, po prostu kliknij na ten link)
- strona pokaże dużą liczbę zbiorów danych SRA (w momencie pisania tego tekstu było ich 2223). To są dane z badania opisującego analizę SARS-CoV-2 w rejonie Bostonu.
- Pobierz metadane opisujące te zbiory danych za pomocą:
- klikając Wyślij do: dropdown
- wybierając
File- zmieniając Format na
RunInfo- klikając Utwórz plikjest to jak powinno wyglądać:
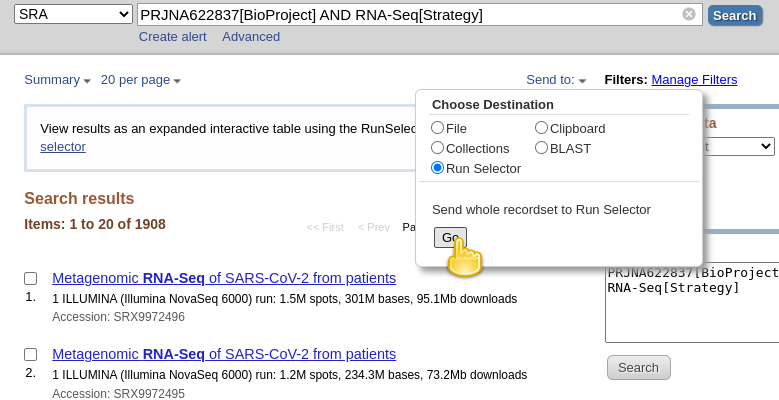
- spowoduje to utworzenie dość dużego pliku
SraRunInfo.csvw folderzeDownloads.
Po pobraniu tego pliku możemy przejść do instancji Galaxy i rozpocząć jej przetwarzanie.
skomentuj komentarz
zauważ, że plik, który właśnie pobraliśmy, nie jest sekwencjonowaniem samych danych. Jest to raczej metadane opisujące właściwości sekwencjonowania odczytów. Przefiltrujemy tę listę do kilku akcesoriów, które będą używane w dalszej części tego samouczka.
proces i filtr SraRunInfo.plik csv w Galaxy
hands_on Hands-on: Upload SraRunInfo.plik csv do galaktyki
- przejdź do wybranej instancji galaktyki, takiej jak jedna z usegalaxy.org, usegalaxy.eu, usegalaxy.org.au ani żadnej innej. (Ten poradnik wykorzystuje usegalaxy.org).
- kliknij przycisk Prześlij dane:
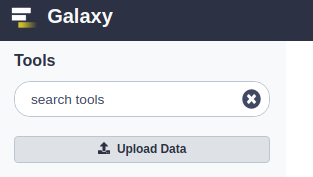
- w oknie dialogowym, które się pojawi, kliknij przycisk „Wybierz pliki lokalne”:

- znajdź i wybierz
SraRunInfo.csvPlik z komputera- kliknij przycisk Start
- Zamknij okno dialogowe, naciskając Zamknij przycisk
- możesz teraz spojrzeć na zawartość tego pliku, klikając ikonę Galaxy-eye (oko). Zobaczysz, że ten plik zawiera wiele informacji na temat poszczególnych akcesji SRA. W badaniu tym każde przystąpienie odpowiada indywidualnemu pacjentowi, którego próbki zostały zsekwencjonowane.
Galaxy może przetwarzać wszystkie 2000+ zbiorów danych, ale aby ten samouczek był znośny, musimy wybrać mniejszy podzbiór. W szczególności nasze wcześniejsze doświadczenia z tymi danymi pokazują dwa interesujące zbiory danych SRR11954102 I SRR12733957. Wyciągnijmy je.
komentarz uwaga na cięcia
sekcja praktyczna poniżej wykorzystuje narzędzie Cut. Istnieją dwa narzędzia cięcia w galaktyce ze względów historycznych. Ten przykład wykorzystuje narzędzie o pełnej nazwie Wytnij kolumny z tabeli (Wytnij). Jednak ta sama logika odnosi się do drugiego narzędzia. Po prostu ma nieco inny interfejs.
hands_on Hands-on: Tworzenie podzbioru danych
- znajdź narzędzie „Wybierz linie pasujące do wyrażenia” narzędzie w sekcji filtruj i sortuj w panelu Narzędzia.
wskazówka: Znajdź narzędzia
Galaxy może mieć zainstalowaną przytłaczającą liczbę narzędzi. Aby znaleźć konkretne narzędzie, wpisz nazwę narzędzia w polu wyszukiwania panelu narzędzia, aby znaleźć narzędzie.
- upewnij się, że
SraRunInfo.csvzbiór danych, który właśnie przesłaliśmy, jest wymieniony w polu param-file „Select lines from” formularza narzędzia.- w polu „wzór” wprowadź następujące wyrażenie →
SRR12733957|SRR11954102. Są to dwa elementy, które chcemy znaleźć oddzielone symbolem potoku||oznaczaor: znajdź wiersze zawierająceSRR12733957lubSRR11954102.- kliknij przycisk
Execute.- to wygeneruje plik zawierający dwie linie (cóż … jedna linia jest również używana jako nagłówek, więc pojawi się, że plik ma trzy linie. Jest OK.)
- Wytnij pierwszą kolumnę z pliku za pomocą narzędzia „Wytnij”, które znajdziesz w sekcji manipulacja tekstem w panelu narzędzi.
- upewnij się, że zbiór danych utworzony w poprzednim kroku jest wybrany w polu „plik do wycięcia” formularza narzędzia.
- Zmień „rozdzielone przez” na
Comma- w „liście pól” wybierz
Column: 1.- Hit
Executespowoduje to utworzenie pliku tekstowego z dwoma wierszami:SRR12733957SRR11954102
teraz, gdy mamy identyfikatory zbiorów danych, które chcemy musimy pobrać dane z sekwencjonowania.
Pobierz dane sekwencjonowania z szybszym pobieraniem i wyciąganiem w FASTQ
hands_on Hands-on: opis zadania
- szybsze pobieranie i wyciąganie odczytuje w narzędziu FASTQ o następujących parametrach:
- „wybierz typ wejścia”:
List of SRA accession, one per line
- parametr param-file „Sra accession list” powinien wskazywać wyjście narzędzia „Cut” z poprzedniego kroku.
- kliknij przycisk
Execute. Spowoduje to uruchomienie narzędzia, które pobiera sekwencje odczytywanych zbiorów danych dla uruchomień wymienionych wSRA. To może trochę potrwać. To może być dobry moment na kawę.- kilka wpisów zostanie utworzonych w panelu historii po przesłaniu tego zadania:
Pair-end data (fasterq-dump): Zawiera sparowane zbiory danych (jeśli są dostępne)Single-end data (fasterq-dump)zawiera pojedyncze zbiory danych (jeśli są dostępne)Other data (fasterq-dump)zawiera Nieparowane zbiory danych (jeśli są dostępne)fasterq-dump logzawiera informacje o wykonanie narzędzia
pierwsze trzy pozycje są w rzeczywistości zbiorami zbiorów danych. Zbiory w galaktyce są logicznymi grupami zbiorów danych, które odzwierciedlają semantyczne relacje między nimi w eksperymencie / analizie. W tym przypadku narzędzie tworzy osobną kolekcję dla sparowanych odczytów końcowych, pojedynczych odczytów i innych.Zobacz tutoriale kolekcji, aby uzyskać więcej informacji.
Przeglądaj Kolekcje, klikając najpierw nazwę kolekcji w panelu Historia. To przeniesie Cię do wnętrza kolekcji i pokaże Ci zbiory danych w niej. Następnie możesz wrócić do zewnętrznego poziomu swojej historii.
gdyfasterq zakończy przesyłanie danych (wszystkie pola są zielone / gotowe), jesteśmy gotowi do ich analizy.
co teraz?
Możesz teraz analizować pobrane dane za pomocą dowolnych narzędzi do analizy sekwencji i przepływów pracy w Galaxy. SRA przechowuje dane dla każdego możliwego rodzaju eksperymentu * – seq.
Jeśli uruchomiłeś ten samouczek, ale pobrałeś zbiory danych, które Cię interesowały, Zobacz resztę biblioteki GTN, aby dowiedzieć się, jak analizować W Galaxy.
Jeśli jednak pobrałeś zbiory danych użyte w powyższych przykładach tego samouczka, jesteś gotowy do uruchomienia analizy wariantu SARS-CoV-2 poniżej.
Analiza zmienności danych sekwencjonowania SARS-Cov-2
w tej części kursu wykonamy wywołanie wariantu i podstawową analizę zbiorów danych pobranych powyżej. Zaczniemy od pobrania sekwencji odniesienia Wuhan-Hu-1 SARS-CoV-2, następnie wykonamy przycinanie adaptera, wyrównanie i wywołanie wariantu, a na koniec przyjrzymy się geograficznemu rozmieszczeniu niektórych znalezionych wariantów.
skomentuj* Projekt analizy COVID-19
Ten poradnik wykorzystuje podzbiór danych i przebiega przez Analizę zmienności covid19.galaxyproject.org.Dane dla covid19.galaxyproject.org jest stale aktualizowana w miarę upubliczniania nowych zbiorów danych.
Pobierz referencyjne dane genomu
obecnie referencyjne dane genomu dotyczą SARS-CoV-2, „Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome”, o ID przystąpienia NC_045512.2.
te dane są dostępne od Zenodo za pomocą poniższego linku.
hands_on Hands-on: Get the reference genome data
Importuj następujący plik do swojej historii:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzWskazówka: Importowanie za pomocą linków
- skopiuj lokalizację linku
- Otwórz Menedżera przesyłania Galaxy (galaxy-upload w prawym górnym rogu panelu Narzędzia)
- wybierz Wklej/Pobierz dane
- wklej link do pola tekstowego
- naciśnij Start
- Zamknij windowprzez domyślnie Galaxy używa adresu URL jako nazwy, więc zmień nazwy plików na bardziej użyteczną nazwę.
przycinanie adapterów za pomocą Fastp
usunięcie adapterów sekwencjonujących poprawia wyrównanie i wywoływanie wariantów. narzędzie fastp może automatycznie wykrywać szeroko stosowane Adaptery sekwencjonowania.
hands_on Hands-On: opis zadania
- narzędzie fastp o następujących parametrach:
- „odczyty jednostronne lub sparowane”:
Paired Collection
- param-file „Select paired collection(s)”:
list_paired(wyjście szybszego pobierania i wyodrębniania czyta się w narzędziu FASTQ)- w „opcjach wyjściowych”:
- „raport wyjściowy JSON”:
Yeswyrównanie z mapą za pomocą BWA-mem
narzędzie BWA-mem jest szeroko stosowanym korektorem sekwencji do krótkich sekwencjonowania zbiorów danych, takich jak te, które analizujemy w tym samouczku.
hands_on Hands-on: Wyrównaj odczyty sekwencjonowania do genomu referencyjnego
- Mapa z narzędziem BWA-MEM O następujących parametrach:
- ” czy wybierzesz Genom referencyjny z historii, czy użyjesz wbudowanego indeksu?”:
Use a genome from history and build index
- param-file” Użyj następującego zestawu danych jako sekwencji odniesienia”:
output(zestaw danych wejściowych)” pojedyncze lub sparowane odczyty końca”:
Paired Collection
- param-file” select a paired Collection”:
output_paired_coll(wyjście narzędzia fastp)- ” Ustaw informacje o odczytanych grupach?”:
Do not set- „Wybierz tryb analizy”:
1.Simple Illumina modeUsuń duplikaty za pomocą MarkDuplicates
narzędzie MarkDuplicates usuwa duplikaty pochodzące z biblioteki przygotowanie artefaktów i sekwencjonowanie artefaktów. Ważne jest, aby usunąć te sekwencje artefaktualne, aby uniknąć sztucznego nadreprezentacji pojedynczej cząsteczki.
hands_on Hands-on: Usuń duplikaty PCR
- narzędzie MarkDuplicates o następujących parametrach:
- param-file „Select sam/BAM dataset or dataset collection”:
bam_output(wyjście mapy za pomocą narzędzia BWA-MEM)- „jeśli prawda nie zapisuje duplikatów do pliku wyjściowego zamiast zapisywać je z ustawionymi odpowiednimi flagami”:
YesGeneruj statystyki wyrównania za pomocą samtools stats
po powtórzeniu powyższego kroku znakowania możemy wygenerować statystyki dotyczące wygenerowanego wyrównania.
hands_on Hands-on: Generowanie statystyk wyrównania
- narzędzie do statystyk Samtools o następujących parametrach:
- param-file „Plik BAM”:
outFile(wyjście narzędzia MarkDuplicates)- „Set coverage distribution”:
No- „Output”:
One single summary file- „Filtruj według flagi sam”:
Do not filter- „użyj sekwencji odniesienia”:
No- „Filtruj według regionów”:
NoRealign odczytuje z lofreq viterbi
Realign odczytuje narzędzie koryguje nieprawidłowe ustawienia wokół wstawiania i usuwania. Jest to wymagane w celu dokładnego wykrycia wariantów.
hands_on Hands-on: Realign odczytuje indele
- Realign odczytuje za pomocą narzędzia lofreq z następującymi parametrami:
- param-file „odczytuje do realign”:
outFile(wyjście narzędzia MarkDuplicates)- „wybierz źródło genomu referencyjnego”:
History
- param-file „Reference”:
output(zestaw danych wejściowych)- w „opcjach zaawansowanych”:
- „Jak obsługiwać podstawowe właściwości 2?”:
Keep unchangedDodaj jakość indel za pomocą lofreq Wstaw jakość indel
ten krok dodaje jakość indel do naszego pliku wyrównania. Jest to konieczne do wywołania wariantów za pomocą narzędzia Lofreq
hands_on Hands-on: Dodaj jakość indel
- Wstaw jakość indel za pomocą narzędzia lofreq o następujących parametrach:
- param-file „Reads”:
realigned(wyjście narzędzia Realign reads)- „Indel calculation approach”:
Dindel
- „Wybierz źródło dla referencyjnego genomu”:
History
- param-file „reference”:
output(zestaw danych wejściowych)warianty wywołania korzystając z opcji wywołania lofreq
jesteśmy teraz gotowi do wywołania wariantów.
hands_on Hands-on: warianty połączeń
- warianty połączeń z narzędziem lofreq o następujących parametrach:
- param-file „input reads in BAM format”:
output(wyjście narzędzia Insert indel)- „Wybierz źródło dla Genom referencyjny”:
History
- param-file „reference”:
output(zestaw danych wejściowych)- „warianty wywołania w poprzek”:
Whole reference- „rodzaje wariantów do wywołania”:
SNVs and indels- „Variant calling parameters”:
Configure settings
- w „Coverage”:
- „Minimal coverage”:
50- w „Base-calling quality”:
- „minimum baseq”:
30- w „quality mapping”:
- „minimum mapping quality”:
20- „variant filter parameters”:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)wyjście tego kroku jest zbiorem plików VCF, które mogą być wizualizowane w przeglądarce genomu.
opisywanie efektów wariantowych za pomocą SnpEff eff:
będziemy teraz opisywać warianty nazwane w poprzednim kroku z efektem, jaki mają na Genom SARS-CoV-2.
hands_on Hands-on: Adnotuj efekty wariantu
- SnpEff eff: narzędzie o następujących parametrach:
- param-file”zmiany sekwencji (SNPs, MNPs, InDels)”:
variants(narzędzie wyjścia wariantów połączeń)- „format wyjściowy”:
VCF (only if input is VCF)- „Utwórz raport CSV, przydatny do dalszej analizy (-csvStats)”:
Yes- „opcje adnotacji”: `
- „filter output”: `
- „filter output specific effects”:
Nowyjście tego kroku jest plikiem VCF z dodanymi efektami wariantu.
Tworzenie tabeli wariantów za pomocą SnpSift Extract Fields
będziemy teraz wybierać różne efekty z VCF i utworzyć plik tabelaryczny, który jest łatwiejszy do zrozumienia dla ludzi.
hands_on Hands-on: Utwórz tabelę wariantów
- narzędzie do wyodrębniania pól SnpSift o następujących parametrach:
- param-file „plik wejściowy wariantu w formacie VCF”:
snpeff_output(wyjście snpeff eff: tool)- „pola do wyodrębnienia”:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- „separator wielu pól”:
,- „pusty tekst pola”:
.możemy sprawdzić pliki wyjściowe i sprawdzić, czy warianty w tym pliku są również opisane w obserwowalnym notatniku, który pokazuje geograficzną dystrybucja sekwencji wariantów SARS-COV-2
interesujące warianty obejmują wariant C do T w pozycji 14408 (14408c/t) w srr11772204, 28144t/c w srr11597145 i 25563g/T w srr11667145.
podsumowanie danych za pomocą MultiQC
podsumujemy teraz naszą analizę za pomocą MultiQC, który generuje piękny raport dla naszych danych.
hands_on Hands-on: podsumowanie danych
- narzędzie MultiQC o następujących parametrach:
- w „Results”:
- param-repeat „Insert Results”
- „które narzędzie zostało użyte do generowania logów?”:
fastp
- param-file „wyjście fastp”:
report_json(wyjście narzędzia fastp)- param-repeat „Insert Results”
- „które narzędzie zostało użyte do generowania logów?”:
Samtools
- In” samtools output”:
- param-repeat” Insert samtools output „
- ” Type of Samtools output?”:
stats
- param-file”Samtools stats output”:
output(wyjście narzędzia Samtools stats)- param-repeat „Insert Results”
- „które narzędzie zostało użyte do generowania logów?”:
Picard
- In” Picard output”:
- param-repeat” Insert Picard output „
- ” Type of Picard output?”:
Markdups- param-file”:
metrics_file(wyjście narzędzia MarkDuplicates)- param-repeat „Insert Results”
- „które narzędzie zostało użyte do generowania logów?”:
SnpEff
- param-file” wyjście SnpEff”:
csvFile(wyjście snpeff eff: narzędzie)wnioski
gratulacje, teraz wiesz, jak importować dane sekwencyjne z sra i jak przeprowadzić przykładową analizę tych zbiorów danych.
punkty kluczowe punkty
dane sekwencji w SRA mogą być bezpośrednio importowane do Galaxy
Często zadawane pytania
masz pytania dotyczące tego samouczka? Sprawdź stronę FAQ na temat analizy wariantu, aby sprawdzić, czy twoje pytanie jest tam wymienione. Jeśli nie, zadaj pytanie na kanale GTN Gitter lub forum Pomocy Galaxy
przydatna Literatura
Więcej informacji, w tym linki do dokumentacji i oryginalnych publikacji, dotyczących narzędzi, technik analizy i interpretacji wyników opisanych w tym poradniku można znaleźć tutaj.
Feedback
czy korzystałeś z tego materiału jako instruktor? Zachęcamy do przekazania nam opinii na temat tego, jak poszło.
powołując się na ten poradnik
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 z archiwum sekwencji odczytu NCBI (Sra) do Galaxy: SARS-analiza wariantu Cov-2 (Materiały szkoleniowe Galaxy). / materiały szkoleniowe / tematy / analiza wariantowa / tutoriale / sars-cov-2 / tutorial.html Online; dostępne już dziś
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016 / j.cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}