formålet med denne tutorial er at introducere dig til behandlingen af næste generations sekventeringsdata i galaksen. Denne tutorial bruger en COVID-19-variant, der ringer fra Illumina data, men det handler ikke om variantopkald i sig selv.
Ved afslutningen af denne tutorial vil du vide:
- Sådan finder du data i SRA og overfører disse oplysninger til galakse
- Sådan udføres grundlæggende NGS-databehandling i galakse inklusive:
- kvalitetskontrol af Illumina data
- kortlægning
- fjernelse af dubletter
- Variant opkald med
lofreq - Variant annotation
- brug af datasætsamlinger
- import af data til Jupyter
### Agenda>> i denne vejledning dækker vi:>1. TOC> {: toc} > {: .agenda} # # to stier gennem denne tutorialvi oprettede to baner, som du kan følge gennem denne tutorial.1. ** Bane 1 * * – start med NCBIS SRA og søg efter tilgængelige tiltrædelser. ** Bane 2* * – omgå NCBI SRA og starte med galaksen direkte. Vi anbefaler at begynde med **Bane 2**.# Sekvensen læst Arkivethe (https://www.ncbi.nlm.nih.gov/sra) er det primære arkiv for *usamlet læser* for (https://www.ncbi.nlm.nih.gov/). SRA er et godt sted at få de sekventeringsdata, der ligger til grund for publikationer og undersøgelser.Denne tutorial dækker, hvordan man får sekvensdata fra SRA ind i galaksen ved hjælp af en direkte forbindelse mellem de to.> ### kommentar kommentar>> du vil også høre SRA kaldet *Short Read Archive*, dets oprindelige navn.> {:.kommentar} # # adgang til SRASRA kan nås enten direkte via dets hjemmeside eller gennem værktøjspanelet på galaksen.> # # # kommentar kommentar>>oprindeligt findes værktøjspanelindstillingen til adgang til SRA kun på (https://usegalaxy.org/). Støtte til den direkte forbindelse til SRA vil blive inkluderet i 20.05 udgivelsen af galakse{: .kommentar} > ### hands_on Hands-on: Udforsk SRA entres>> 1. Gå til din galakse forekomst af valg som en af (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) eller enhver anden. (Denne tutorial bruger usegalaxy.org).> 1. Hvis din historie ikke allerede er tom, end starte en ny historie (Se (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) for mere om galakse historier)> 1. ** Klik på * * ‘Hent Data’ øverst i værktøjspanelet.> 1. ** Klik * * ‘Sra Server `i listen over værktøjer vist under`Hent Data’.>dette tager dig (https://www.ncbi.nlm.nih.gov/sra) – du kan også starte direkte fra SRA. Et søgefelt vises øverst på siden. Prøv at søge efter noget, du er interesseret i, såsom `delfin` eller `nyre` eller `delfin nyre` og derefter **klik på** knappen `Søg`.>> dette returnerer en liste over *Sra-eksperimenter*, der matcher din søgestreng. Sra-eksperimenter, også kendt som *SRK-poster*, indeholder sekvensdata fra et bestemt eksperiment samt en forklaring af selve eksperimentet og andre relaterede data. Du kan udforske de returnerede eksperimenter ved at klikke på deres navn. Se (https://www.ncbi.nlm.nih.gov/bøger/NBK56913/) i (https://www.ncbi.nlm.nih.gov/bøger/n/helpsrakb/) for mere.>>når du indtaster tekst i Sra-søgefeltet, bruger du (https://www.ncbi.nlm.nih.gov/sra /docs/srasearch/). Det understøtter både enkle tekstsøgninger og meget præcise søgninger, der kontrollerer specifikke metadata og bruger vilkårligt komplekse logiske udtryk. Det giver dig mulighed for at skalere dine søgninger fra grundlæggende til avanceret, når du indsnævrer dine søgninger. Syntaksen for avancerede søgninger kan virke skræmmende, men SRA giver en grafisk (https://www.ncbi.nlm.nih.gov/sra/avanceret/) for at generere den specifikke syntaks. Og som vi skal se nedenfor, giver SRA Run Selector en endnu venligere brugergrænseflade til at indsnævre vores valgte data.>> spil rundt med Sra-Entresgrænsefladen, herunder den avancerede forespørgselsbygger, for at se om du kan identificere et sæt SRA-eksperimenter, der er relevante for et af dine forskningsområder.{: .hands_on} > # # # hands_on Hands-on: Generer liste over matchende eksperimenter ved hjælp af entres>> nu hvor du har en grundlæggende fortrolighed med SRA entres, lad os finde de sekvenser, der bruges i denne tutorial.>> 1. Hvis du ikke allerede er der, **Naviger * * tilbage til (https://www.ncbi.nlm.nih.gov/sra> 1. ** Ryd * * enhver søgetekst fra søgefeltet.> 1. ** Skriv * * ‘sars-cov-2 `i søgefeltet og **klik på**`Søg’.> dette returnerer en lang liste over SRA-eksperimenter, der matcher vores søgning, og denne liste er alt for lang til at bruge i en tutorial-øvelse. På dette tidspunkt kunne vi bruge den avancerede forespørgselsbygger, vi lærte om ovenfor.> men det gør vi ikke. lad os i stedet sende *for længe til en tutorial* liste resultater, vi har til Sra Run-vælgeren, og brug dens venligere grænseflade til at indsnævre vores resultater.>> !(../../ billeder / sra_center.png){: .hands_on} > # # # hands_on Hands-on: Gå fra entres til Sra Run Selector>> se resultater som en udvidet interaktiv tabel ved hjælp af RunSelector.>> 1. Klik på Send resultater for at køre vælgeren, som vises i et felt øverst i søgeresultaterne.>> !(../../ billeder / sra_centr_resultat.png)>>> ### tip hvad hvis du ikke kan se Kørevælgerlinket?>>>> du har måske bemærket denne tekst tidligere, da du udforskede Entresøgning. Denne tekst vises kun noget af tiden, når antallet af søgeresultater falder inden for et ret bredt vindue. Du kan ikke se det, hvis du kun har et par resultater, og du kan ikke se det, hvis du har flere resultater, end Kørevælgeren kan acceptere.>>>> *du skal komme til at køre vælgeren for at sende dine resultater til galaksen.* Hvad hvis du ikke har nok resultater til at udløse dette link bliver vist? I så fald ringer du til Kørevælgeren ved at **klikke** på rullemenuen `Send til` øverst til højre i resultatpanelet. For at komme til Run Selector,* * Vælg * * ‘Run Selector `og derefter** klik på * * knappen` Go’.> !(../../ billeder / sra_centr_send_to.png)> {: .tip} >>> 1. ** Klik på* * `Send resultater til Kørevælger ‘ øverst i søgeresultatpanelet. (Hvis du ikke kan se dette link, så se kommentaren direkte ovenfor.){: .hands_on} # # Sra Run Selectorvi lærte tidligere, hvordan vi indsnævrer vores søgeresultater ved hjælp af entres avancerede syntaks. Vi udnyttede dog ikke den magt, da vi var i Entree. I stedet brugte vi en simpel søgning og sendte derefter alle resultaterne til Kørevælgeren. Vi har endnu ikke den (korte) liste over resultater, vi vil køre analyse på. * Hvad laver vi?* Brug Startgrænsefladen til at indsnævre dine resultater til en størrelse, som Kørevælgeren kan forbruge. * Brug Kørevælgerens meget venligere grænseflade til 1. Forstå lettere de data, vi har 1. Begræns disse resultater ved hjælp af denne viden.> ### kommentar Kørevælger er både mere og mindre end entres>> Kørevælger kan gøre mest, men ikke alt hvad Entresøgningssyntaks kan gøre. Run selector bruger * facetteret søgning * teknologi, som er nem at bruge, og kraftfuld, men som har iboende grænser. Specifikt fungerer entres bedre, når du søger på attributter, der har tiere, hundreder eller tusinder af forskellige værdier. Kør Selector vil fungere bedre søge attributter med færre end 20 forskellige værdier. Heldigvis beskriver det de fleste søgninger.{: .kommentar}vinduet Kørevælger er opdelt i flere paneler:* **`filterliste`**: i øverste venstre hjørne. Det er her, vi vil forfine vores søgning.* * * ‘Vælg’**: en oversigt over, hvad der oprindeligt blev sendt til Run Selector, og hvor meget af det, vi har valgt indtil videre. (Og indtil videre har vi ikke valgt noget af det.) Bemærk også den fristende, men stadig gråtonede `galakse` – knap. I første omgang er dette listen over elementer, der sendes til Kørevælgeren. Denne liste krymper, når vi anvender filtre på den.!(../../ billeder / sra_run_selector.png)> ### kommentar Hvorfor gik antallet af fundne varer *op?*>> Husk, at entres-grænsefladen viser Sra-eksperimenter. Kør Selector lister * kører * — sekventering datasæt-og der er* en eller flere * kører pr eksperiment. Vi har de samme data som før, vi ser det nu bare i finere detaljer.{: .kommentar} ‘filterlisten’ øverst til venstre viser kolonner i vores resultater, der enten har kontinuerlige numeriske værdier eller 10 eller mindre (du kan ændre dette tal) forskellige værdier i dem. ** Rul* * ned gennem listen Vælg et par af filtrene. Når et filter er valgt, vises en* værdier * boks nedenfor, notering muligheder for dette filter, og antallet af kørsler med hver indstilling. Disse værdier / indstillinger trækkes fra datasætmetadataene. Prøv * * valg* * et par interessante klingende filtre og derefter **vælg** en eller flere muligheder for hvert filter. Prøv * * fravælg * * indstillinger og filtre. Når du gør dette, vil antallet af fundne resultater falde eller stige.> # # # tip Tip: Brug filtre til bedre at forstå dataene>>filtre er, hvordan du indsnævrer de datasæt, der overvejes til afsendelse til galakse, men de er også en glimrende måde at forstå dine data på:> for det første er valg af et filter en nem måde at se værdiområdet i en kolonne. Du kan muligvis ikke (https://www.google.com/search?q=sra+sirs_outcome), men du kan muligvis finde ud af det ved at se, hvilke værdier der er i det.> for det andet kan du undersøge, hvordan forskellige kolonner forholder sig til hinanden. Er der en sammenhæng mellem `sirs_outcome` værdier og `disease_stage` værdier?{: .tip}> ### hands_on Hands-on: Begræns dine resultater ved hjælp af Run Selector>> 1. Hvis du har nogen filtre tændt, **Fravælg * * dem.> når du har gjort dette, vises der ikke nogen *værdier* – bokse under `filterlisten`.> 2. ** Kopier og indsæt * * denne søgestreng i søgefeltet ‘fundne emner’.>> SRR11772204 eller SRR11597145 eller SRR11667145>> denne hånd-plukket sæt kørsler begrænser vores resultater til 3 kørsler fra forskellige geografiske fordeling.{: .hands_on}dette reducerer din liste over` fundne varer ‘ fra titusinder af kørsler til 3 kørsler (et håndterbart nummer til en tutorial!). Men vi er ikke helt færdige med Run Selector endnu. Bemærk, at knappen` galakse ‘ stadig er nedtonet. Vi har indsnævret vores muligheder, men vi har faktisk ikke valgt noget at sende til galaksen endnu.Det er muligt at vælge hvert resterende løb ved at **klikke på** afkrydsningsfeltet øverst i den første kolonne. Du kan fravælge alt ved at * * klikke på * * * `K`.> ### hands_on Hands-on: vælg kørsler og send til galakse>> 1. Vælg alle kørsler ved at * * klikke* * på`K’.> og nu er `galakse` knappen levende.> 1. ** Klik på * * knappen` galakse ‘i afsnittet` Vælg’ øverst på siden.{: .hands_on} # # tilbage i Galaksenår vi klikker på ‘galakse’ i Kørevælgeren, sker der flere ting. Først, det lancerer en ny fane eller et vindue, der åbnes i galaksen. Du vil se den * store grønne boks*, der angiver, at håndtrykket mellem SRA og galaksen var vellykket, og du vil så se et nyt `Sra` job i dit historiepanel. Denne boks kan starte som grå / verserende, hvilket indikerer, at overførslen endnu ikke er startet, eller den kan gå direkte til gul / løbende eller til grøn / færdig.> # # # hands_on Hands-on: Undersøg det nye Sra-datasæt>> 1. Når overførslen ‘ Sra ‘ er afsluttet, **klik** på datasætets galakse-øje (øje) ikon.>> dette viser datasættet i galaksens centerpanel.{: .hands_on}` SRA ‘ datasættet er ikke sekvensdata, men snarere *metadata*, som vi vil bruge til at hente sekvensdata fra Sra. Denne metadata afspejler de oplysninger, vi så I Kørevælgerens `fundne elementer` sektion. Metadataene er ikke de slutdata, vi søger fra SRA, men at have alle disse metadata er ofte nyttige i efterfølgende analysetrin.Lad os nu bruge disse metadata til at hente sekvensdata fra Sra. SRA giver værktøjer til udvinding af alle former for information, herunder selve sekvensdataene. Det er baseret på Sra (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump) nytte, og gør netop det.– >
- Find nødvendige data i Sra
- hands_on Hands-on: opgavebeskrivelse
- kommentar kommentar
- proces og filter SraRunInfo.csv-fil i galaksen
- hands_on Hands-on: Upload SraRunInfo.CSV-fil i galaksen
- kommentar pas på nedskæringer
- hands_on Hands-on: oprettelse af en delmængde af data
- tip Tip: Find værktøjer
- Hent sekventeringsdata med hurtigere hentning og uddrag læser i fastk
- hands_on Hands-on: opgavebeskrivelse
- hvad nu?
- Variationsanalyse af SARS-Cov-2 sekventeringsdata
- kommenter usegalaksen.* COVID-19 analyseprojekt
- Hent referencegenomdataene
- hands_on Hands-on: Hent referencegenomdataene
- Tip: import via links
- Adapter trimning med fastp
- hands_on Hands-on: opgavebeskrivelse
- justering med kort med BVA-mem
- hands_on Hands-on: Juster sekventering læser at referere genom
- Fjern dubletter med MarkDuplicates
- hands_on Hands-on: Fjern PCR dubletter
- generer justeringsstatistik med samtools statistik
- hands_on Hands-on: Generer justeringsstatistik
- Juster læser værktøj korrigerer fejljusteringer omkring indsættelser og sletninger. Dette er nødvendigt for nøjagtigt at registrere varianter. hands_on Hands-on: Relign læser omkring indels Relign læser med lofrek-værktøj med følgende parametre: param-file “Reads to relign”: outFile (output af MarkDuplicates tool) “vælg kilden til referencegenomet”: History param-file “Reference”: output (Input datasæt) i” Avancerede indstillinger”: ” Hvordan håndteres basiskvaliteter af 2?”: Keep unchanged Tilføj indel kvaliteter med Lofrek Indsæt indel kvaliteter
- hands_on Hands-on: Tilføj Indel kvaliteter
- opkaldsvarianter brug af opkaldsvarianter
- hands_on Hands-on: Opkaldsvarianter
- Kommenter varianteffekter med SnpEff eff:
- hands_on Hands-on: Kommenter varianteffekter
- Opret tabel med varianter ved hjælp af SnpSift-Ekstraktfelter
- hands_on Hands-on: Opret tabel over varianter
- Opsummer data med Multicc
- hands_on Hands-on: opsummere data
- konklusion
- keypoints nøglepunkter
- Ofte Stillede Spørgsmål
- nyttig litteratur
- Feedback
- citerer denne Tutorial
- details BibTeX
Find nødvendige data i Sra
først skal vi finde et godt datasæt at lege med. Sekvenslæsearkivet (SRA) er det primære arkiv for ikke-samlede læsninger, der drives af US National Institutes of Health (NIH). SRA er et godt sted at få de sekventeringsdata, der ligger til grund for publikationer og undersøgelser. Lad os gøre det:
hands_on Hands-on: opgavebeskrivelse
- gå til NCBI ‘ s SRA-side ved at pege din bro. ser til https://www.ncbi.nlm.nih.gov/sra
- i søgefeltet indtast
SARS-CoV-2 Patient Sequencing From Partners / MGH(alternativt skal du blot klikke på dette link)
- hjemmesiden viser et stort antal SRA-datasæt (i skrivende stund var der 2.223). Dette er data fra en undersøgelse, der beskriver analyse af SARS-CoV-2 i Boston-området.
- Hent metadata, der beskriver disse datasæt ved:
- klik på Send til: rullemenu
- valg af
File- ændring af Format til
RunInfo- Klik på Opret filher er hvordan det skal se ud:
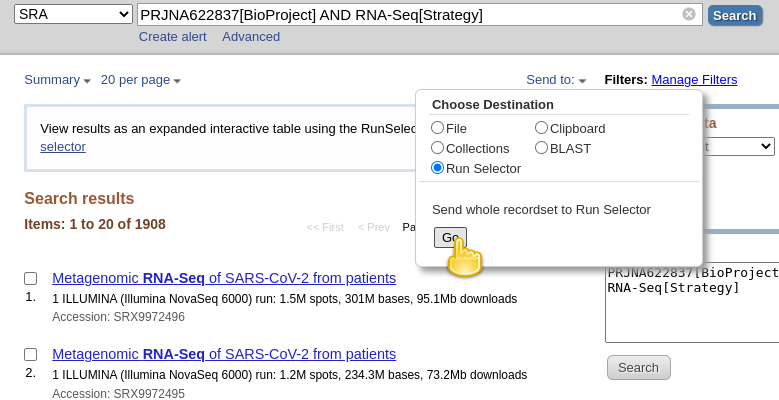
- dette ville skabe en ret stor
SraRunInfo.csvfil i dinDownloadsmappe.
nu hvor vi har hentet denne fil, kan vi gå til en Galakseinstans og begynde at behandle den.
kommentar kommentar
Bemærk, at den fil, vi lige har hentet, ikke sekventerer data selv. Det er snarere metadata, der beskriver egenskaber ved sekventering læser. Vi filtrerer denne liste ned til blot et par tiltrædelser, der vil blive brugt i resten af denne tutorial.
proces og filter SraRunInfo.csv-fil i galaksen
hands_on Hands-on: Upload SraRunInfo.CSV-fil i galaksen
- gå til din galakse forekomst af valg, såsom en af usegalaxy.org, usegalaxy.eu, usegalaxy.org.au eller noget andet. (Denne tutorial bruger usegalaxy.org).
- Klik på knappen Upload Data:
- i dialogboksen, der vises, skal du klikke på knappen “Vælg lokale filer”:

- Find og vælg
SraRunInfo.csvfil fra din computer- Klik på startknappen
- Luk dialog ved at trykke på Luk knappen
- du kan nu se på indholdet af denne fil ved at klikke på galakse-øje (øje) ikon. Du vil se, at denne fil indeholder en masse oplysninger om individuelle Sra tiltrædelser. I denne undersøgelse svarer hver tiltrædelse til en individuel patient, hvis prøver blev sekventeret.
galakse kan behandle alle 2.000+ datasæt, men for at gøre denne tutorial tålelig skal vi vælge en mindre delmængde. Især vores tidligere erfaring med disse data viser to interessante datasæt SRR11954102og SRR12733957. Så lad os trække dem ud.
kommentar pas på nedskæringer
det praktiske afsnit nedenfor bruger skæreværktøj. Der er to skæreværktøjer i galaksen på grund af historiske årsager. Dette eksempel bruger værktøj med det fulde navn klip kolonner fra en tabel (klip). Den samme logik gælder dog for det andet værktøj. Det har simpelthen en lidt anden grænseflade.
hands_on Hands-on: oprettelse af en delmængde af data
- Find værktøj “Vælg linjer, der matcher et udtryk” værktøj i Filter og sorter sektion af værktøjspanelet.
tip Tip: Find værktøjer
galakse kan have en overvældende mængde værktøjer installeret. For at finde et bestemt værktøj skal du skrive værktøjsnavnet i værktøjspanelets søgefelt for at finde værktøjet.
- sørg for, at
SraRunInfo.csvdatasæt, vi lige har uploadet, er angivet i feltet param-file “Select lines from” I værktøjsformularen.- i feltet “mønster” skal du indtaste følgende udtryk Kris
SRR12733957|SRR11954102. Dette er to tiltrædelser, vi ønsker at finde adskilt af rørsymbolet||betyderor: find linjer indeholdendeSRR12733957ellerSRR11954102.- Klik på
Executeknap.- dette genererer en fil, der indeholder to linjer (Godt … En linje bruges også som overskrift, så den vises filen har tre linjer. Det er OK.)
- klip den første kolonne fra filen ved hjælp af værktøjet “klip” værktøj, som du finder i Tekstmanipulationsafsnittet i værktøjsruden.
- sørg for, at datasættet, der er produceret af det foregående trin, er valgt i feltet “File to cut” i værktøjsformularen.
- Skift “afgrænset af” til
Comma- i” liste over felter”vælg
Column: 1.- Hit
Executedette vil producere en tekstfil med kun to linjer:SRR12733957SRR11954102
nu hvor vi har identifikatorer af datasæt, har vi en tekstfil, der er vil vi nødt til at hente de faktiske sekventering data.
Hent sekventeringsdata med hurtigere hentning og uddrag læser i fastk
hands_on Hands-on: opgavebeskrivelse
- hurtigere hentning og uddrag læser i fastk værktøj med følgende parametre:
- “select input type”:
List of SRA accession, one per line
- parameteren param-fil” Sra tiltrædelsesliste “skal pege på udgangen af værktøjet” Cut ” fra det foregående trin.
- Klik på knappen
Execute. Dette vil køre værktøjet, som henter sekvensen læse datasæt for de kørsler, der blev opført iSRAdatasæt. Det kan tage lidt tid. Så dette kan være et godt tidspunkt at få kaffe.- flere poster oprettes i dit historikpanel, når du sender dette job:
Pair-end data (fasterq-dump): Indeholder parrede datasæt (hvis tilgængelige)Single-end data (fasterq-dump)indeholder Single-end datasæt (hvis tilgængelige)Other data (fasterq-dump)indeholder uparrede datasæt (hvis tilgængelige)fasterq-dump logindeholder oplysninger om værktøjets udførelse
de første tre elementer er faktisk samlinger af datasæt. Samlinger i galaksen er logiske grupperinger af datasæt, der afspejler de semantiske forhold mellem dem i eksperimentet / analysen. I dette tilfælde opretter værktøjet en separat samling hver for parrede slutlæsninger, enkeltlæsninger og andet.Se samlinger tutorials for mere.
Udforsk samlingerne ved først at klikke på samlingsnavnet i panelet Historik. Dette tager dig inde i samlingen og viser dig datasættene i den. Du kan derefter navigere tilbage til det ydre niveau af din historie.
Når fasterq afslutter overførsel af data (alle kasser er grønne / færdige), er vi klar til at analysere det.
hvad nu?
Du kan nu analysere de hentede data ved hjælp af alle sekvensanalyseværktøjer og arbejdsgange i galaksen. SRA har sikkerhedskopieringsdata for enhver tænkelig type *-seks eksperiment.
Hvis du kørte denne tutorial, men hentede datasæt, som du var interesseret i, så se resten af GTN-biblioteket for ideer til, hvordan man analyserer i galakse.
men hvis du hentede de datasæt, der blev brugt i denne vejlednings eksempler ovenfor, er du klar til at køre SARS-CoV-2-variantanalysen nedenfor.
Variationsanalyse af SARS-Cov-2 sekventeringsdata
i denne del af vejledningen udfører vi variantopkald og grundlæggende analyse af datasæt, der er hentet ovenfor. Vi starter med at hente SARS-cov-2-referencesekvensen, derefter køre adaptertrimning, justering og variantopkald og endelig se på den geografiske fordeling af nogle af de fundne varianter.
kommenter usegalaksen.* COVID-19 analyseprojekt
denne tutorial bruger en delmængde af dataene og løber gennem variationsanalyseafsnittet af covid19.galakseprojekt.org.Data for covid19.galaxyproject.org opdateres løbende, efterhånden som nye datasæt offentliggøres.
Hent referencegenomdataene
referencegenomdataene for i dag er for SARS-CoV-2, “alvorligt akut respiratorisk syndrom coronavirus 2 isolat Vuhan-Hu-1, komplet genom”, der har tiltrædelses-ID for NC_045512.2.
disse data er tilgængelige via følgende link.
hands_on Hands-on: Hent referencegenomdataene
Importer følgende fil i din historie:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzTip: import via links
- Kopier linkplaceringen
- Åbn galakse Upload Manager (galakse-upload øverst til højre på værktøjspanelet)
- vælg Indsæt/hent data
- indsæt linket i tekstfeltet
- tryk på Start
- luk vinduetgalakse bruger som standard URL ‘ en som navn, så omdøb filerne med et mere nyttigt navn.
Adapter trimning med fastp
fjernelse af sekventeringsadaptere forbedrer justeringer og variantopkald. fastp værktøj kan automatisk registrere udbredte sekventering adaptere.
hands_on Hands-on: opgavebeskrivelse
- fastp-værktøj med følgende parametre:
- “Single-end eller paired reads”:
Paired Collection
- param-file ” Select paired collection (s)”:
list_paired(output af hurtigere hentning og uddrag læser i hurtig værktøj)- i” Output Options”:
- ” Output JSON report”:
Yesjustering med kort med BVA-mem
BVA-mem-værktøj er en meget brugt sekvensjustering til kortlæste sekvensdatasæt som dem, vi analyserer i denne tutorial.
hands_on Hands-on: Juster sekventering læser at referere genom
- kort med BVA-mem værktøj med følgende parametre:
- “vil du vælge en reference genom fra din historie eller bruge en indbygget indeks?”:
Use a genome from history and build index
- param-file “Brug følgende datasæt som referencesekvens”:
output(Input datasæt)- “Single or Paired-end reads”:
Paired Collection
- param-File “vælg en parret samling”:
output_paired_coll(output af fastp tool)- “Indstil læse grupper information?”:
Do not set- “Select analysis mode”:
1.Simple Illumina modeFjern dubletter med MarkDuplicates
MarkDuplicates tool fjerner dublerede sekvenser, der stammer fra biblioteket forberedelse artefakter og sekventering artefakter. Det er vigtigt at fjerne disse artefaktuelle sekvenser for at undgå kunstig overrepræsentation af enkelt molekyle.
hands_on Hands-on: Fjern PCR dubletter
- MarkDuplicates værktøj med følgende parametre:
- param-file “vælg SAM/BAM datasæt eller datasætsamling”:
bam_output(output af kort med BVA-mem-værktøj)- “hvis sandt skriv ikke duplikater til outputfilen i stedet for at skrive dem med passende flag sæt”:
Yesgenerer justeringsstatistik med samtools statistik
efter det duplikerede markeringstrin ovenfor kan vi generere statistik om den justering, vi har genereret.
hands_on Hands-on: Generer justeringsstatistik
- Samtools stats tool med følgende parametre:
- param-file “BAM file”:
outFile(output af MarkDuplicates tool)- “Set coverage distribution”:
No- “Output”:
One single summary file- “Filtrer efter Sam-flag”:
Do not filter- “brug en referencesekvens”:
No- “Filtrer efter regioner”:
NoJuster læser værktøj korrigerer fejljusteringer omkring indsættelser og sletninger. Dette er nødvendigt for nøjagtigt at registrere varianter.
hands_on Hands-on: Relign læser omkring indels
- Relign læser med lofrek-værktøj med følgende parametre:
- param-file “Reads to relign”:
outFile(output af MarkDuplicates tool)- “vælg kilden til referencegenomet”:
History
- param-file “Reference”:
output(Input datasæt)- i” Avancerede indstillinger”:
- ” Hvordan håndteres basiskvaliteter af 2?”:
Keep unchangedTilføj indel kvaliteter med Lofrek Indsæt indel kvaliteter
dette trin tilføjer indel kvaliteter i vores justeringsfil. Dette er nødvendigt for at kalde varianter ved hjælp af Opkaldsvarianter med lofrek-værktøj
hands_on Hands-on: Tilføj Indel kvaliteter
- Indsæt Indel kvaliteter med lofrek værktøj med følgende parametre:
- param-fil “læser”:
realigned(output af Relign læser værktøj)- “Indel beregning tilgang”:
Dindel
- “Vælg kilden til referencegenomet”:
History
- param-file “reference”:
output(input datasæt)opkaldsvarianter brug af opkaldsvarianter
Vi er nu klar til at ringe til varianter.
hands_on Hands-on: Opkaldsvarianter
- Opkaldsvarianter med lofrek-værktøj med følgende parametre:
- param-file “Input læser i BAM-format”:
output(output af Indsæt Indel-kvalitetsværktøj)- “Vælg kilden til referencegenomet”:
History
- param-file “reference”:
output(input datasæt)- “opkaldsvarianter på tværs af”:
Whole reference- “typer af varianter, der skal ringes til”:
SNVs and indels- “Variant calling parameters”:
Configure settings
- i “Coverage”:
- “Minimal coverage”:
50- i “Base-calling”:
- “minimum baseks”:
30- “minimum baseks for alternative baser”:
30- i “kortlægningskvalitet
20- “variantfilterparametre”:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
udgangen af dette trin er en samling af VCF-filer, der kan visualiseres i en genom-bro.ser.
Kommenter varianteffekter med SnpEff eff:
Vi vil nu kommentere de varianter, vi kaldte i det foregående trin med den effekt, de har på SARS-CoV-2 genomet.
hands_on Hands-on: Kommenter varianteffekter
- SnpEff eff: værktøj med følgende parametre:
- param-file “Sekvensændringer (SNPs, MNPs, InDels)”:
variants(output of Call variants tool)- “outputformat”:
VCF (only if input is VCF)- “Opret CSV-rapport, nyttig til nedstrømsanalyse (- csvStats)”:
Yes- “Annotationsindstillinger”: `
- “filteroutput”: `
- “filtrer specifikke effekter”:
No
udgangen af dette trin er en VCF-fil med tilføjede varianteffekter.
Opret tabel med varianter ved hjælp af SnpSift-Ekstraktfelter
Vi vælger nu forskellige effekter fra VCF og opretter en tabelfil, der er lettere at forstå for mennesker.
hands_on Hands-on: Opret tabel over varianter
- SnpSift Uddrag felter værktøj med følgende parametre:
- param-file “Variant input file in VCF format”:
snpeff_output(output af SnpEff eff: tool)- “felter til uddrag”:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- “multiple field separator”:
,- “tom felttekst”:
.
Vi kan inspicere outputfilerne og se kontrollere, om varianter i denne fil også er beskrevet i en observerbar notesbog, der viser de geografisk fordeling af SARS-cov-2 variantsekvenser
interessante varianter inkluderer C til t-varianten i position 14408 (14408c/t) i srr11772204, 28144t/C i srr11597145 og 25563g/t i srr11667145.
Opsummer data med Multicc
vi vil nu opsummere vores Analyse med Multicc, som genererer en smuk rapport for vores data.
hands_on Hands-on: opsummere data
- Multicc værktøj med følgende parametre:
- I “resultater”:
- param-gentag “Indsæt resultater”
- “hvilket værktøj blev brugt generer logfiler?”:
fastp
- param-fil “Output af fastp”:
report_json(output af fastp tool)- param-gentag “Indsæt resultater”
- “hvilket værktøj blev brugt generer logfiler?”:
Samtools
- i “Samtools output”:
- param-gentag” Indsæt Samtools output “
- ” Type Samtools output?”:
stats
- param-fil “Samtools stats output”:
output(output af Samtools stats tool)- param-gentag “Indsæt resultater”
- “hvilket værktøj blev brugt generer logfiler?”:
Picard
- i “Picard output”:
- param-gentag” Indsæt Picard output “
- ” Type Picard output?”:
Markdups- param-fil “Picard output”:
metrics_file(output af MarkDuplicates tool)- param-gentag “Indsæt resultater”
- “hvilket værktøj blev brugt generer logfiler?”:
SnpEff
- param-file “Output af SnpEff”:
csvFile(output af SnpEff eff: værktøj)
konklusion
tillykke, du ved nu, hvordan du importerer sekvensdata fra SRA, og hvordan du kører en eksempelanalyse på disse datasæt.
keypoints nøglepunkter
sekvensdata i SRA kan importeres direkte til galaksen
Ofte Stillede Spørgsmål
har du spørgsmål om denne tutorial? Tjek siden Ofte stillede spørgsmål for Variantanalyseemnet for at se, om dit spørgsmål er angivet der. Hvis ikke, bedes du stille dit spørgsmål på GTN Gitterkanalen eller galaksens Hjælpeforum
nyttig litteratur
yderligere information, herunder links til dokumentation og originale publikationer, vedrørende værktøjer, analyseteknikker og fortolkning af resultater beskrevet i denne vejledning kan findes her.
Feedback
brugte du dette materiale som instruktør? Du er velkommen til at give os feedback om, hvordan det gik.
citerer denne Tutorial
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekratenko, 2021 fra NCBI ‘ s sekvens Læs arkiv (Sra) til galakse: SARS-CoV-2 variant analyse (galakse træningsmaterialer). / uddannelse-materiale/emner/variant-analyse/tutorials/sars-cov-2 / tutorial.html Online; adgang til i dag
- Batut et al., 2018 samfundsdrevet Dataanalysetræning til Biologicellesystemer 10.1016 / j.cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}