aminosyrer, nukleotider eller enhver anden evolutionær karakter erstattes afandre med en vis hastighed. Forestil dig for eksempel en evolutionær sekvens med tremulige tilstande, A, B og C. Hvis substitutionsmodellen er tidsomvendelig, vil der være tre overgangshastigheder, a<>B, B<>C og a<>C.
Antag, at satserne er henholdsvis 1, 1 og 0 i substitutionsenheder pr.100tegn pr. tidsenhed. Efter en tidsenhed, i en 300 tegn lang sekvens oprindeligt sammensat ligeligt af As, Bs og Cs, forventer vi, at der skal være en A til B-substitution og en b til C-substitution. Hvis vi sammenligner to homologe sekvenser i levende organismer, fordi en tidsenhed er gået for begge sekvenser, ville vi forvente to a til B og to B til Csubstitutioner mellem nutidens sekvenser.
uanset hvor længe vi kører denne proces, vil der aldrig være en direkte udskiftning af A med C. Der vil heller aldrig være en A til C-substitution under aso-kaldet infinite sites model, hvor der ikke kan forekomme mere end en substitution på et enkelt sted.
men da A til B og B til C substitutioner er almindelige, under en endelig sitesmodel til sidst B vil blive erstattet af C på et sted, hvor A tidligere blev erstattet af B. Denne indirekte udskiftning af A med C (eller tilsvarende i atime-reversibel model, C med a) bliver mere sandsynlig, jo længere tidsperiodenseparation af de homologe sekvenser.
Jeg simulerede sekvensudvikling baseret på ovenstående scenario, kører thesimulation i 10 tidsenheder. Fra denne substitution observerede jeg følgende tællinger for hvert sted mønster:
| A | B | C | |
|---|---|---|---|
| A | 91 | 9 | 0 |
| B | 5 | 86 | 9 |
| C | 0 | 9 | 91 |
inden for denne relativt korte varighed ser det ikke ud som om nogen a<>csubstitutioner har fundet sted. Men når jeg reran simuleringen for 100 unitsof tid:
| A | B | C | |
|---|---|---|---|
| A | 55 | 35 | 10 |
| B | 29 | 36 | 35 |
| C | 20 | 36 | 44 |
som du kan se, er mange “a” – tegn blevet erstattet med “C” og viceversa. Mere generelt, under en endelig lokalitetsmodel flere substitutionerforårsage, at fordelingen af antallet af lokalitetsmønstre bliver meget fladere ud over blot at øge andelen af off-diagonal i forhold til diagonale tællinger.Pam og BLOSUM score matricer tegner sig for flere substitutioner iradikalt forskellige måder.
PAM-matricerne for aminosyrer sammen med forkortelserne med et enkelt bogstavanvendes til genetisk kodede aminosyrer, blev udviklet af MargaretDayhoff. De blev oprindeligt udgivet i 1978 og baseret på proteinsekvenserne, som Dayhoff havde samlet siden 1960 ‘ erne, udgivet som theAtlas af proteinsekvens og struktur.
navnet PAM kommer fra” punkt accepteret mutation ” og henviser tiludskiftning af en enkelt aminosyre i et protein med en anden aminosyre.Disse mutationer blev identificeret ved at sammenligne meget lignende sekvenser med mindst 85% identitet, og det antages, at eventuelle observerede substitutioner var resultatet af en enkelt mutation mellem forfædresekvensen og en af de nuværende dags sekvenser.
PAM definerer også en tidsenhed, hvor 1 PAM er den tid, hvor 1/100 aminoacider forventes at gennemgå en mutation. PAM1-sandsynlighedsmatricen viser sandsynligheden for, at aminosyren ved kolonne j erstattes af aminoacidet i række i. det blev beregnet ud fra Dayhoffs PAM-tællinger, og omskaleret tobe 1 PAM tidsenhed. Som du kan se, er de off-diagonale sandsynligheder ipam1-matricen alle meget små (alle elementer blev skaleret med 10.000 forlegbarhed):
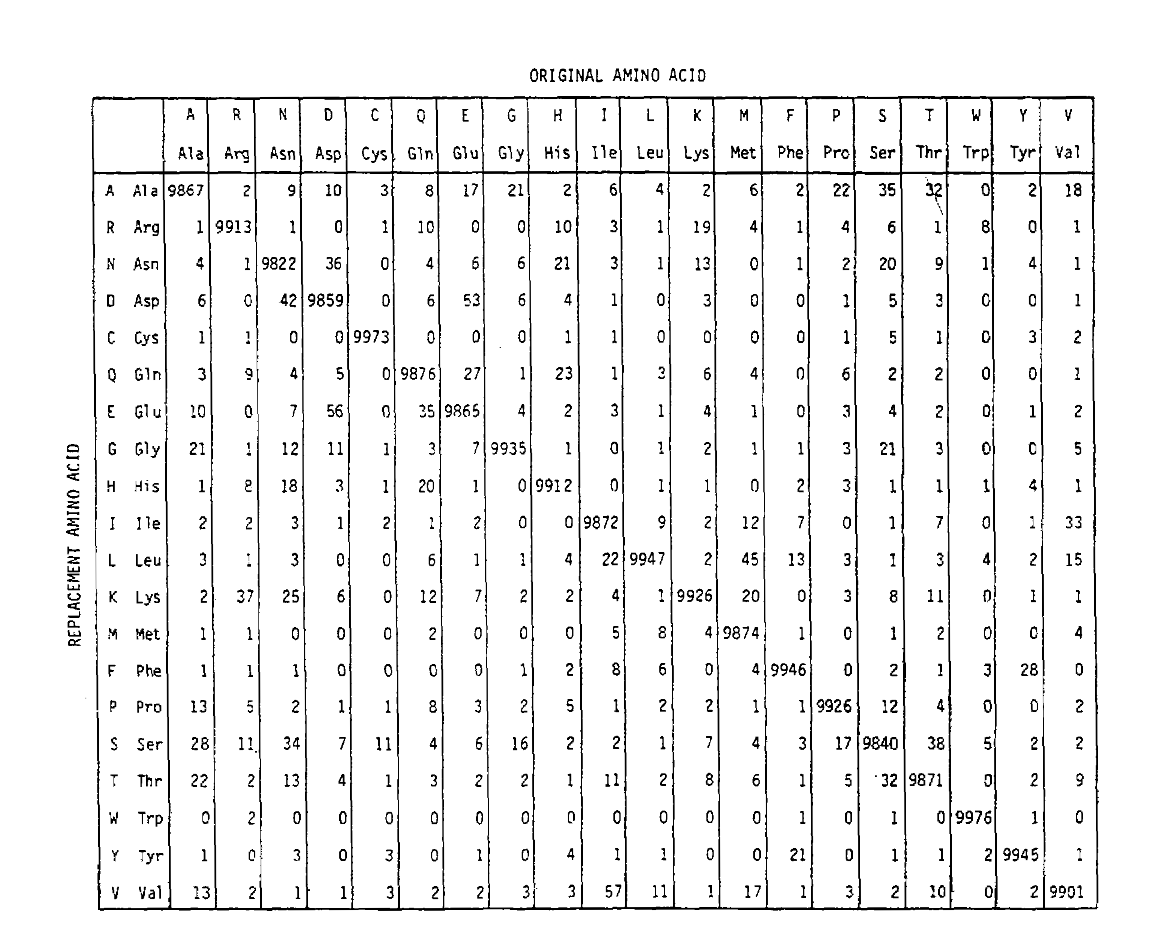
for at beregne aminosyreudskiftningssandsynlighederne i længere tiddurationer kan matricen multipliceres med sig selv det tilsvarende antal gange. Således blev PAM250 sandsynlighedsmatricen, der beskriver udskiftningssandsynlighederne givet 250 PAM tidsenheder, afledt ved at hæve PAM1 sandsynlighedsmatricen til effekten 250 (alle elementer blev skaleret med 100for læsbarhed):
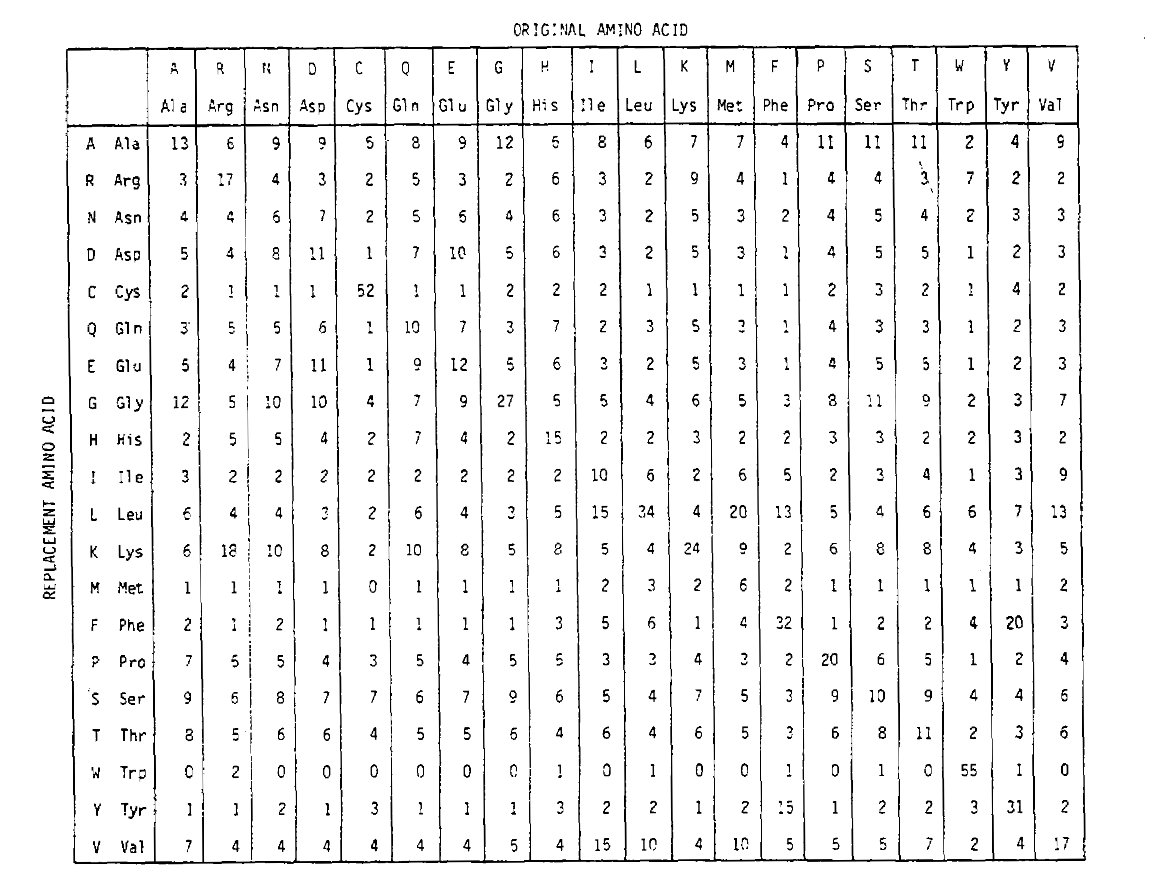
udskiftningssandsynlighederne afledt ved hjælp af denne eksponentiering korrektregnskab for flere substitutioner. Ikke alene er off-diagonalsandsynlighederne forholdsmæssigt større, som du ville forvente i længere tidvarighed, men de er fladere. For eksempel er sandsynligheden for en valin (V)til isoleucin (I) udskiftning 33 liter større end en V til histadin (H)erstatning i PAM1-matricen, men kun 4,5 liter større i PAM250-matricen.
Score matricer kan derefter beregnes ud fra sandsynlighedsmatricer ogobserverede basefrekvenser.
BLOSUM matricer, udviklet af Steven og Jorja Henikoff og udgivet i1992, tager en meget anden tilgang. Mens PAM implicit anvender en stationær endelig lokalitetsmodel for evolution ved hjælp af matrice eksponentiering, behandles effekten af flere substitutioner implicit i BLOSUM ved at konstruere forskellige score matricer til forskellige tidsskalaer.
inden for flere sekvensjusteringer af homologe sekvenser identificeres konserverede sammenhængende blokke af aminosyrer. Inden for hver blok grupperes multisekvenser, når deres parvise gennemsnitlige sekvensidentitet er højere end en tærskel. Tærsklen er 80% for BLOSUM80-matricen, 62% for BLOSUM62, 50% for BLOSUM50 og så videre.
dette betyder, at for BLOSUM80 vil blokke have gennemsnitlige parvise identiteter, der ikke er større end 80%, for BLOSUM62 ikke større end 62% osv.
Aminosyreudskiftningssandsynligheder for homologe sekvenser beregnesfra parvise sammenligninger mellem klynger. Disse sandsynligheder vil være resultatet af enkelt-og multiple substitutioner, hvor flere substitutioner har større indflydelse på større evolutionære afstande. Derfor vil scorematricer genereret fra parvise sammenligninger mellem klynger med gennemsnitlig større afstand, som BLOSUM50-matricen, naturligvis redegøre for den større effekt af flere substitutioner.
selvom de tager forskellige ruter, er de endelige blosum og PAM score matricerer faktisk ret ens. Ifølge Henikoff og Henikoff er følgende Pam-og BLOSUM-matricer sammenlignelige:
| PAM | BLOSUM |
|---|---|
| PAM250 | BLOSUM45 |
| PAM160 | BLOSUM62 |
| PAM120 | BLOSUM80 |
For more information on PAM (Dayhoff) and BLOSUM matrices, see kapitel 2 afbiologisk sekvensanalyse af Durbin et al. og Facebook.
opdatering 13. oktober 2019: for et andet perspektiv på substitutionsmatricer, se afsnittet “omveje” i slutningen af kapitel 5 i Bioinformatikalgoritmer (2.eller 3. udgave) af Compeau og Pevsner.