Le but de ce tutoriel est de vous présenter le traitement des données de séquençage de nouvelle génération dans Galaxy. Ce tutoriel utilise un appel de variante COVID-19 à partir des données Illumina, mais il ne s’agit pas d’un appel de variante en soi.
À la fin de ce tutoriel, vous saurez:
- Comment trouver des données dans SRA et transférer ces informations dans Galaxy
- Comment effectuer un traitement de données NGS de base dans Galaxy, y compris:
- Contrôle qualité (QC) des données Illumina
- Mappage
- Suppression des doublons
- Appel de variantes avec
lofreq - Annotation de variantes
- Utilisation de collections de jeux de données
- Importation de données dans Jupyter
### Agenda >>Dans ce tutoriel, nous allons couvrir: >> 1. TOC > {:toc}> {: .agenda } ## Deux chemins à travers ce didacticielnous avons créé deux trajectoires que vous pouvez suivre à travers ce tutoriel.1. ** Trajectoire 1 ** – commencez par le SRA de NCBI et recherchez les accessions disponibles → Start (#the-sequence-read-archive) 2. ** Trajectoire 2 ** – contournez le SRA de NCBI et commencez directement par Galaxy. → Démarrer (# retour dans la galaxie) Nous vous recommandons de commencer par ** Trajectoire 2 **.# L’archive de lecture de la séquence (https://www.ncbi.nlm.nih.gov/sra) est l’archive primaire de *lectures non assemblées * pour le (https://www.ncbi.nlm.nih.gov/). SRA est un excellent endroit pour obtenir les données de séquençage qui sous-tendent les publications et les études.Ce tutoriel explique comment obtenir des données de séquence de SRA dans Galaxy en utilisant une connexion directe entre les deux.> ###commentaire Commentaire >>Vous entendrez également SRA appelée *Archive de lecture courte*, son nom d’origine.> {:.commentaire } ## L’accès à SRASRA peut être atteint soit directement via son site Web, soit via le panneau d’outils sur Galaxy.> ###commentaire Commentaire >>Initialement, l’option du panneau d’outils pour accéder à SRA n’existe que sur le (https://usegalaxy.org/). La prise en charge de la connexion directe à SRA sera incluse dans la version 20.05 de Galaxy {:.comment} > ###hands_on Hands-on: Explorez SRA Entrez >>1. Accédez à votre instance Galaxy de choix, telle que l’une des (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) ou toute autre. (Ce tutoriel utilise usegalaxy.org ). > 1. Si votre historique n’est pas déjà vide, commencez un nouvel historique (voir (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) pour en savoir plus sur les historiques de galaxies) >1. ** Cliquez sur ** « Obtenir des données » en haut du panneau d’outils.> 1. ** Cliquez sur ** ‘Serveur SRA’ dans la liste des outils affichés sous ‘Obtenir des données’.> Cela vous prend le (https://www.ncbi.nlm.nih.gov/sra) you vous pouvez également démarrer directement à partir du SRA. Une zone de recherche est affichée en haut de la page. Essayez de rechercher quelque chose qui vous intéresse, comme « dauphin » ou « rein » ou « rein de dauphin », puis ** cliquez ** sur le bouton « Rechercher ».>> Cela renvoie une liste d’expériences *SRA* qui correspondent à votre chaîne de recherche. Les expériences SRA, également appelées entrées *SRX*, contiennent des données de séquence d’une expérience particulière, ainsi qu’une explication de l’expérience elle-même et de toute autre donnée connexe. Vous pouvez explorer les expériences retournées en cliquant sur leur nom. Voir (https://www.ncbi.nlm.nih.gov/books/NBK56913/) dans le (https://www.ncbi.nlm.nih.gov/books/n/helpsrakb/) pour plus d’informations.>>Lorsque vous entrez du texte dans la zone de recherche SRA, vous utilisez (https://www.ncbi.nlm.nih.gov/sra/docs/srasearch/). Entrez prend en charge à la fois des recherches de texte simples et des recherches très précises qui vérifient des métadonnées spécifiques et utilisent des expressions logiques arbitrairement complexes. Entrez vous permet d’étendre vos recherches de base à avancées au fur et à mesure que vous affinez vos recherches. La syntaxe des recherches avancées peut sembler intimidante, mais SRA fournit un graphique (https://www.ncbi.nlm.nih.gov/sra/advanced/) pour générer la syntaxe spécifique. Et, comme nous le verrons ci-dessous, le sélecteur d’exécution SRA fournit une interface utilisateur encore plus conviviale pour réduire nos données sélectionnées.>> Jouez avec l’interface SRA Entrez, y compris le générateur de requêtes avancé, pour voir si vous pouvez identifier un ensemble d’expériences SRA pertinentes pour l’un de vos domaines de recherche.{: .hands_on}> ###hands_on Pratique: Générer une liste d’expériences correspondantes en utilisant Entrez >> Maintenant que vous avez une connaissance de base de SRA Entrez, trouvons les séquences utilisées dans ce tutoriel.>> 1. Si vous n’y êtes pas déjà, ** revenez ** au (https://www.ncbi.nlm.nih.gov/sra>1. ** Effacer ** tout texte de recherche de la zone de recherche.> 1. ** Tapez ** ‘sars-cov-2’ dans la zone de recherche et ** cliquez sur ** ‘Rechercher’.> Cela renvoie une longue liste d’expériences SRA qui correspondent à notre recherche, et cette liste est beaucoup trop longue pour être utilisée dans un exercice de tutoriel. À ce stade, nous pourrions utiliser le générateur de requêtes Entre avancé que nous avons appris ci-dessus.> Mais nous ne le ferons pas. À la place, envoyons les résultats de la liste * trop longs pour un tutoriel * que nous avons au sélecteur d’exécution SRA, et utilisons son interface plus conviviale pour affiner nos résultats.>>!(../../ images / sra_entrez.png) {:.hands_on}> ###hands_on Pratique: Passez de Entrez au sélecteur d’exécution SRA >> Affichez les résultats sous la forme d’une table interactive étendue à l’aide du sélecteur d’exécution.>> 1. Cliquez sur Envoyer les résultats pour Exécuter le sélecteur, qui apparaît dans une boîte en haut des résultats de recherche.>>!(../../ images/sra_entrez_result.png) >>> ### astuce Que se passe-t-il si vous ne voyez pas le lien du sélecteur d’exécution?>>>>Vous avez peut-être remarqué ce texte plus tôt lorsque vous exploriez Entrer search. Ce texte n’apparaît qu’une partie du temps, lorsque le nombre de résultats de recherche tombe dans une fenêtre assez large. Vous ne le verrez pas si vous n’avez que quelques résultats, et vous ne le verrez pas si vous avez plus de résultats que ce que le sélecteur d’exécution peut accepter.>>>>* Vous devez exécuter Selector pour envoyer vos résultats à Galaxy.* Que se passe-t-il si vous n’avez pas assez de résultats pour déclencher l’affichage de ce lien? Dans ce cas, vous appelez accéder au sélecteur d’exécution en cliquant *** sur le menu déroulant `Envoyer à` en haut à droite du panneau de résultats. Pour accéder au sélecteur d’exécution, ** sélectionnez ** ‘Sélecteur d’exécution’ puis ** cliquez sur ** le bouton `Go’.>!(../../ images / sra_entrez_send_to.png) > {: .tip}>>> 1. ** Cliquez sur ** « Envoyer les résultats au sélecteur d’exécution » en haut du panneau des résultats de recherche. (Si vous ne voyez pas ce lien, consultez le commentaire directement ci-dessus.){: .hands_on} ## SRA Run Selectornous avons appris plus tôt comment affiner nos résultats de recherche en utilisant la syntaxe avancée d’Entrez. Cependant, nous n’avons pas profité de ce pouvoir lorsque nous étions dans Entrez. Au lieu de cela, nous avons utilisé une recherche simple, puis envoyé tous les résultats au sélecteur d’exécution. Nous n’avons pas encore la (courte) liste des résultats sur lesquels nous voulons exécuter l’analyse. * Que faisons-nous?* Nous utilisons Entre et le Sélecteur d’exécution comment ils sont conçus pour être utilisés: * Utilisez l’interface Entre pour réduire vos résultats à une taille que le Sélecteur d’exécution peut consommer. * Envoyez ces résultats d’Entrée au Sélecteur d’exécution SRA * Utilisez l’interface beaucoup plus conviviale du sélecteur d’exécution à 1. Comprendre plus facilement les données dont nous disposons 1. Limitez ces résultats en utilisant ces connaissances.> ### le sélecteur d’exécution de commentaires est à la fois supérieur et inférieur à Entre >> Le sélecteur d’exécution peut faire la plupart, mais pas tout ce que la syntaxe de recherche Entre peut faire. Le sélecteur d’exécution utilise * la technologie de recherche à facettes * qui est facile à utiliser et puissante, mais qui a des limites inhérentes. Plus précisément, Entrez fonctionnera mieux lors de la recherche sur des attributs qui ont des dizaines, des centaines ou des milliers de valeurs différentes. Le sélecteur d’exécution fonctionnera mieux avec les attributs de recherche avec moins de 20 valeurs différentes. Heureusement, cela décrit la plupart des recherches.{: .commentaire} La fenêtre du sélecteur d’exécution est divisée en plusieurs panneaux : *** `Liste des filtres` **: Dans le coin supérieur gauche. C’est là que nous affinerons notre recherche.*** ‘Select’ **: Un résumé de ce qui a été initialement transmis à Run Selector et de la quantité que nous avons sélectionnée jusqu’à présent. (Et jusqu’à présent, nous n’en avons sélectionné aucune.) Notez également le bouton alléchant, mais toujours grisé, « Galaxy ».*** `Éléments trouvés x` ** Initialement, il s’agit de la liste des éléments envoyés au sélecteur d’exécution à partir d’Entre. Cette liste se rétrécira au fur et à mesure que nous y appliquerons des filtres.!(../../ images / sra_run_sélecteur.png) > ### commentaire Pourquoi le nombre d’objets trouvés* a-t-il augmenté?*>> Rappelons que l’interface Entrez répertorie les expériences SRA (entrées SRX). Listes de sélecteurs d’exécution *runs— – séquençage des ensembles de données — et il y a * un ou plusieurs *runs par expérience. Nous avons les mêmes données qu’auparavant, nous les voyons maintenant plus en détail.{: .commentaire} La « liste des filtres » en haut à gauche affiche des colonnes dans nos résultats qui ont soit des valeurs numériques continues, soit 10 valeurs distinctes ou moins (vous pouvez modifier ce nombre). ** Faites défiler ** vers le bas dans la liste, sélectionnez quelques filtres. Lorsqu’un filtre est sélectionné, une case *valeurs* apparaît ci-dessous, répertoriant les options de ce filtre et le nombre d’exécutions avec chaque option. Ces valeurs/options sont extraites des métadonnées de l’ensemble de données. Essayez ** de sélectionner ** quelques filtres de sondage intéressants, puis ** sélectionnez ** une ou plusieurs options pour chaque filtre. Essayez de désélectionner les options et les filtres. Au fur et à mesure, le nombre de résultats trouvés diminuera ou augmentera.> ### tip tip: Utilisez des filtres pour mieux comprendre les données >>Les filtres permettent de réduire les ensembles de données à envoyer à Galaxy, mais ils sont également un excellent moyen de comprendre vos données: > Tout d’abord, la sélection d’un filtre est un moyen facile de voir la plage de valeurs dans une colonne. Vous ne pouvez peut-être pas le faire (https://www.google.com/search?q=sra+sirs_outcome), mais vous pouvez éventuellement le comprendre en voyant quelles sont les valeurs qu’il contient.>Deuxièmement, vous pouvez explorer comment les différentes colonnes se rapportent les unes aux autres. Existe-t-il une relation entre les valeurs `sirs_outcome` et les valeurs `disease_stage` ?{: .tip} > ###hands_on Hands-on: Affinez vos résultats à l’aide du sélecteur d’exécution >> 1. Si des filtres sont activés, ** désélectionnez-les **.> Une fois que vous avez fait cela, aucune case *valeurs* n’apparaîtra sous la `Liste des filtres`.>2. ** Copiez et collez ** cette chaîne de recherche dans la zone de recherche « Éléments trouvés ».>>SRR11772204 OU SRR11597145 OU SRR11667145 >> Cette main – l’ensemble de pistes choisi limite nos résultats à 3 pistes de distribution géographique différente.{: .hands_on} Cela réduit votre liste d’éléments trouvés de dizaines de milliers d’exécutions à 3 exécutions (un nombre gérable pour un tutoriel!). Mais nous n’en avons pas encore fini avec le sélecteur d’exécution. Notez que le bouton « Galaxy » est toujours grisé. Nous avons réduit nos options, mais nous n’avons pas encore sélectionné quoi que ce soit à envoyer à Galaxy.Il est possible de sélectionner chaque exécution restante en cliquant sur la coche en haut de la première colonne. Vous pouvez tout désélectionner en ** cliquant ** sur le ‘X’.> ###hands_on Hands-on: Sélectionnez les exécutions et envoyez-les à Galaxy >>1. Sélectionnez toutes les exécutions en cliquant sur le » X « .> Et maintenant, le bouton `Galaxy’ est en direct.> 1. ** Cliquez ** sur le bouton « Galaxy » dans la section « Sélectionner » en haut de la page.{: .hands_on } # # De retour dans Galaxylorsque nous cliquons sur `Galaxy` dans le sélecteur d’exécution, plusieurs choses se produisent. Tout d’abord, il lance un nouvel onglet ou une nouvelle fenêtre de navigateur qui s’ouvre dans Galaxy. Vous verrez la *grande boîte verte * indiquant que la poignée de main entre SRA et Galaxy a réussi et vous verrez alors un nouveau travail « SRA » dans votre panneau d’historique. Cette case peut commencer en gris / en attente, indiquant que le transfert n’a pas encore commencé, ou elle peut passer directement au jaune / en cours d’exécution ou au vert / terminé.> ###hands_on Pratique: Examinez le nouvel ensemble de données SRA >>1. Une fois le transfert `SRA’ terminé, ** cliquez ** sur l’icône galaxy-eye (œil) de l’ensemble de données.>> Cela affiche l’ensemble de données dans le panneau central de Galaxy.{: .hands_on} L’ensemble de données `SRA’ n’est pas des données de séquence, mais plutôt des *métadonnées* que nous utiliserons pour obtenir des données de séquence de SRA. Ces métadonnées reflètent les informations que nous avons vues dans la section « Éléments trouvés » du sélecteur d’exécution. Les métadonnées ne sont pas les données finales que nous recherchons auprès de SRA, mais avoir toutes ces métadonnées est souvent utile lors des étapes d’analyse ultérieures.Utilisons maintenant ces métadonnées pour récupérer les données de séquence de SRA. SRA fournit des outils pour extraire toutes sortes d’informations, y compris les données de séquence elles-mêmes. L’outil Galaxy ‘Téléchargement et extraction plus rapides des lectures dans FASTQ’ est basé sur l’utilitaire SRA (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump), et fait exactement cela.– >
- Trouvez les données nécessaires dans SRA
- hands_on Hands-on: Description de la tâche
- commentaire Commentaire
- Traiter et filtrer SraRunInfo.fichier csv dans Galaxy
- hands_on Hands-on: Téléchargez SraRunInfo.fichier csv dans Galaxy
- commentaireAttention aux coupures
- hands_on Hands-on: Création d’un sous-ensemble de données
- astuce Astuce: Outils de recherche
- Télécharger les données de séquençage avec des Lectures de Téléchargement et d’extraction plus rapides dans FASTQ
- hands_on Hands-on:Description de la tâche
- Et maintenant ?
- Analyse des variations des données de séquençage du SARS-Cov-2
- commentez L’utilisation.* Projet d’analyse COVID-19
- Obtenir les données du génome de référence
- hands_on Hands-on: Obtenez les données du génome de référence
- Astuce: Importer via des liens
- Le rognage de l’adaptateur avec fastp
- hands_on Hands-on: Description de la tâche
- Alignement avec la carte avec BWA-MEM
- hands_on Pratique: Aligner les lectures de séquençage sur la carte du génome de référence
- Supprimer les doublons avec MarkDuplicates
- hands_on Hands-on: Supprime les doublons PCR
- Générer des statistiques d’alignement avec Samtools stats
- hands_on Pratique: Générer des statistiques d’alignement
- Réaligner les lectures avec lofreq viterbi
- hands_on Hands-on: Réaligner les lectures autour des indels
- Ajoutez des qualités indel avec lofreq Insérez des qualités indel
- hands_on Hands-on: Ajouter des qualités indel
- Variantes d’appel en utilisant des variantes d’appel lofreq
- hands_on Hands-on: Variantes d’appel
- Annoter les effets des variantes avec SnpEff eff:
- hands_on Hands-on: Annoter les effets de variantes
- Créer une table de variantes à l’aide des champs d’extraction SnpSift
- hands_on Hands-on: Créer une table de variantes
- Résumer les données avec MultiQC
- hands_on Hands-on: Résumez les données
- Conclusion
- points clés Points clés
- Foire aux questions
- Littérature utile
- Feedback
- Citant ce tutoriel
- details BibTeX
Trouvez les données nécessaires dans SRA
Nous devons d’abord trouver un bon jeu de données avec lequel jouer. La Sequence Read Archive (SRA) est la principale archive de lectures non assemblées exploitée par les Instituts nationaux de la santé des États-Unis (NIH). SRA est un excellent endroit pour obtenir les données de séquençage qui sous-tendent les publications et les études. Faisons cela:
hands_on Hands-on: Description de la tâche
- Accédez à la page SRA de NCBI en pointant votre navigateur sur https://www.ncbi.nlm.nih.gov/sra
- Dans la zone de recherche, entrez
SARS-CoV-2 Patient Sequencing From Partners / MGH(Sinon, il vous suffit de cliquer sur ce lien)
- La page Web affichera un grand nombre d’ensembles de données SRA (au moment de la rédaction, il y en avait 2 223). Il s’agit des données d’une étude décrivant l’analyse du SARS-CoV-2 dans la région de Boston.
- Téléchargez les métadonnées décrivant ces ensembles de données en :
- en cliquant sur Envoyer à: liste déroulante
- En sélectionnant
File- En changeant le format en
RunInfo- En cliquant sur Créer un fichieril y a à quoi cela devrait ressembler:
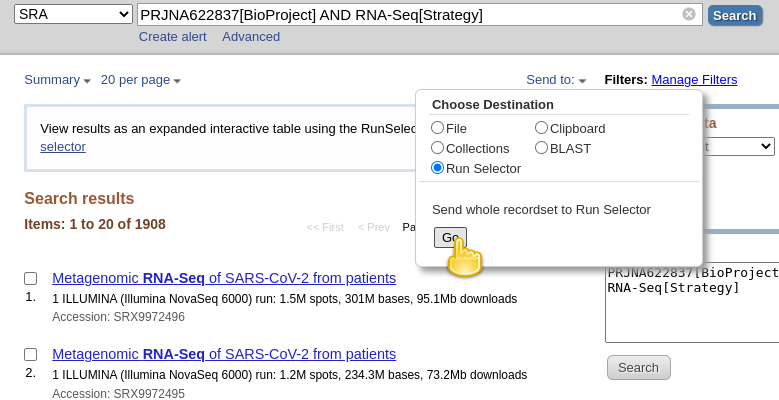
- Cela créerait un fichier
SraRunInfo.csvassez volumineux dans votre dossierDownloads.
Maintenant que nous avons téléchargé ce fichier, nous pouvons accéder à une instance de Galaxy et commencer à le traiter.
commentaire Commentaire
Notez que le fichier que nous venons de télécharger n’est pas le séquençage des données lui-même. Il s’agit plutôt de métadonnées décrivant les propriétés des lectures de séquençage. Nous allons filtrer cette liste en quelques accessions qui seront utilisées dans le reste de ce tutoriel.
Traiter et filtrer SraRunInfo.fichier csv dans Galaxy
hands_on Hands-on: Téléchargez SraRunInfo.fichier csv dans Galaxy
- Accédez à votre instance Galaxy de choix telle que l’une des usegalaxy.org , usegalaxy.eu , usegalaxy.org.au ou tout autre. (Ce tutoriel utilise usegalaxy.org ).
- Cliquez sur le bouton Télécharger les données:
- Dans la boîte de dialogue qui apparaît, cliquez sur le bouton « Choisir les fichiers locaux »:

- Recherchez et sélectionnez
SraRunInfo.csvfichier de votre ordinateur- Cliquez sur le bouton Démarrer
- Fermez la boîte de dialogue en appuyant sur Bouton de fermeture
- Vous pouvez maintenant regarder le contenu de ce fichier en cliquant sur l’icône galaxy-eye (œil). Vous verrez que ce fichier contient beaucoup d’informations sur les adhésions individuelles au SRA. Dans cette étude, chaque accession correspond à un patient individuel dont les échantillons ont été séquencés.
Galaxy peut traiter plus de 2 000 jeux de données, mais pour rendre ce tutoriel supportable, nous devons sélectionner un sous-ensemble plus petit. En particulier, notre expérience précédente avec ces données montre deux ensembles de données intéressants SRR11954102 et SRR12733957. Alors, sortons-les.
commentaireAttention aux coupures
La section pratique ci-dessous utilise l’outil de coupe. Il existe deux outils de coupe dans Galaxy pour des raisons historiques. Cet exemple utilise un outil avec le nom complet Couper les colonnes d’une table (couper). Cependant, la même logique s’applique à l’autre outil. Il a simplement une interface légèrement différente.
hands_on Hands-on: Création d’un sous-ensemble de données
- Outil de recherche « Sélectionnez les lignes qui correspondent à une expression » dans la section Filtrer et trier du panneau d’outils.
astuce Astuce: Outils de recherche
Galaxy peut avoir une quantité écrasante d’outils installés. Pour trouver un outil spécifique, tapez le nom de l’outil dans la zone de recherche du panneau d’outils pour trouver l’outil.
- Assurez-vous que l’ensemble de données
SraRunInfo.csvque nous venons de télécharger est répertorié dans le champ « Sélectionner des lignes dans » du formulaire d’outil.- Dans le champ « le motif », entrez l’expression suivante →
SRR12733957|SRR11954102. Ce sont deux adhésions que nous voulons trouver séparées par le symbole de tuyau|. Le|signifieor: recherche des lignes contenantSRR12733957ouSRR11954102.- Cliquez sur le bouton
Execute.- Cela générera un fichier contenant deux lignes (enfin one une ligne est également utilisée comme en-tête, il apparaîtra donc que le fichier a trois lignes. C’est BON.)
- Coupez la première colonne du fichier à l’aide de l’outil « Couper », que vous trouverez dans la section Manipulation de texte du volet d’outils.
- Assurez-vous que l’ensemble de données produit par l’étape précédente est sélectionné dans le champ « Fichier à couper » du formulaire d’outil.
- Remplacez « Délimité par » par
Comma- Dans « Liste des champs », sélectionnez
Column: 1.- Hit
ExecuteCela produira un fichier texte avec seulement deux lignes:SRR12733957SRR11954102
Maintenant que nous avons des identifiants d’ensembles de données que nous voulons nous devons télécharger les données de séquençage réelles.
Télécharger les données de séquençage avec des Lectures de Téléchargement et d’extraction plus rapides dans FASTQ
hands_on Hands-on:Description de la tâche
- Télécharger et Extraire plus rapidement les Lectures dans l’outil FASTQ avec les paramètres suivants:
- « sélectionner le type d’entrée »:
List of SRA accession, one per line
- Le fichier de paramètres « liste d’adhésion sra » doit pointer la sortie de l’outil « Couper » de l’étape précédente.
- Cliquez sur le bouton
Execute. Cela exécutera l’outil, qui récupère les ensembles de données de lecture de séquence pour les exécutions répertoriées dans l’ensemble de donnéesSRA. Cela peut prendre un certain temps. Donc, c’est peut-être le bon moment pour prendre un café.- Plusieurs entrées sont créées dans votre panneau d’historique lorsque vous soumettez ce travail :
Pair-end data (fasterq-dump): Contient des ensembles de données d’extrémité appariés (si disponibles)Single-end data (fasterq-dump)Contient des ensembles de données d’extrémité unique (si disponibles)Other data (fasterq-dump)Contient des ensembles de données non appariés (si disponibles)fasterq-dump logContient des informations sur l’exécution de l’outil li>
Les trois premiers éléments sont en fait des collections d’ensembles de données. Les collections dans Galaxy sont des regroupements logiques d’ensembles de données qui reflètent les relations sémantiques entre eux dans l’expérience /l’analyse. Dans ce cas, l’outil crée une collection distincte pour chacune des lectures de fin appariées, des lectures simples et autres.Consultez les tutoriels sur les collections pour en savoir plus.
Explorez les collections en cliquant d’abord sur le nom de la collection dans le panneau historique. Cela vous emmène à l’intérieur de la collection et vous montre les jeux de données qu’elle contient. Vous pouvez ensuite revenir au niveau externe de votre historique.
Une fois que fasterq a terminé le transfert des données (toutes les cases sont vertes / terminées), nous sommes prêts à les analyser.
Et maintenant ?
Vous pouvez désormais analyser les données récupérées à l’aide de tous les outils et flux de travail d’analyse de séquence dans Galaxy. SRA détient des données de sauvegarde pour tous les types imaginables d’expériences *-seq.
Si vous avez exécuté ce tutoriel, mais que vous avez récupéré des ensembles de données qui vous intéressaient, consultez le reste de la bibliothèque GTN pour des idées sur la façon d’analyser dans Galaxy.
Cependant, si vous avez récupéré les jeux de données utilisés dans les exemples de ce tutoriel ci-dessus, vous êtes prêt à exécuter l’analyse de la variante SARS-CoV-2 ci-dessous.
Analyse des variations des données de séquençage du SARS-Cov-2
Dans cette partie du tutoriel, nous effectuerons un appel de variantes et une analyse de base des jeux de données téléchargés ci-dessus. Nous allons commencer par télécharger la séquence de référence Wuhan-Hu-1 SARS-CoV-2, puis exécuter le rognage de l’adaptateur, l’alignement et l’appel de variantes et enfin examiner la distribution géographique de certaines des variantes trouvées.
commentez L’utilisation.* Projet d’analyse COVID-19
Ce tutoriel utilise un sous-ensemble des données et passe par la section Analyse de la variation du covid19.projet galaxyproject.org.Les données pour covid19.galaxyproject.org est mis à jour en permanence à mesure que de nouveaux ensembles de données sont rendus publics.
Obtenir les données du génome de référence
Les données du génome de référence pour aujourd’hui sont pour le SARS-CoV-2, « Isolat du coronavirus du syndrome respiratoire aigu sévère 2 Wuhan-Hu-1, génome complet », ayant l’ID d’adhésion de NC_045512.2.
Ces données sont disponibles auprès de Zenodo en utilisant le lien suivant.
hands_on Hands-on: Obtenez les données du génome de référence
Importez le fichier suivant dans votre historique:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzAstuce: Importer via des liens
- Copiez l’emplacement du lien
- Ouvrez le Gestionnaire de téléchargement Galaxy (téléchargement Galaxy en haut à droite du panneau d’outils)
- Sélectionnez Coller/Récupérer Data
- Collez le lien dans le champ de texte
- Appuyez sur Démarrer
- Fermez la fenêtrepar défaut, Galaxy utilise l’URL comme nom, renommez donc les fichiers avec un nom plus utile.
Le rognage de l’adaptateur avec fastp
La suppression des adaptateurs de séquençage améliore les alignements et les appels de variantes. l’outil fastp peut détecter automatiquement les adaptateurs de séquençage largement utilisés.
hands_on Hands-on: Description de la tâche
- outil fastp avec les paramètres suivants:
- « Lectures à une extrémité ou appariées »:
Paired Collection
- fichier param « Sélectionnez la(les) collection(s) appariée(s) »:
list_paired(sortie de Lectures de Téléchargement et d’extraction plus rapides dans l’outil FASTQ)- Dans « Options de sortie »:
- « Rapport JSON de sortie »:
Yes
Alignement avec la carte avec BWA-MEM
L’outil BWA-MEM est un aligneur de séquences largement utilisé pour les ensembles de données de séquençage à lecture courte tels que ceux que nous analysons dans ce tutoriel.
hands_on Pratique: Aligner les lectures de séquençage sur la carte du génome de référence
- avec l’outil BWA-MEM avec les paramètres suivants:
- « Allez-vous sélectionner un génome de référence dans votre historique ou utiliser un index intégré? »:
Use a genome from history and build index
- param-file »Utilise l’ensemble de données suivant comme séquence de référence »:
output(Jeu de données d’entrée)- « Lectures à extrémité unique ou appariée »:
Paired Collection
- param-file « Sélectionnez une collection appariée »:
output_paired_coll(sortie de l’outil fastp)- » Définir les informations des groupes de lecture? »:
Do not set- « Sélectionner le mode d’analyse »:
1.Simple Illumina mode
Supprimer les doublons avec MarkDuplicates
L’outil MarkDuplicates supprime les séquences en double provenant de la préparation de la bibliothèque artefacts et artefacts de séquençage. Il est important de supprimer ces séquences artefactuelles pour éviter la surreprésentation artificielle d’une seule molécule.
hands_on Hands-on: Supprime les doublons PCR
- Outil MarkDuplicates avec les paramètres suivants:
- param-file « Sélectionnez l’ensemble de données SAM /BAM ou la collection d’ensembles de données »:
bam_output(sortie de la carte avec l’outil BWA-MEM)- « Si true, n’écrivez pas de doublons dans le fichier de sortie au lieu de les écrire avec le jeu d’indicateurs approprié »:
Yes
Générer des statistiques d’alignement avec Samtools stats
Après l’étape de marquage en double ci-dessus, nous pouvons générer des statistiques sur l’alignement que nous avons généré.
hands_on Pratique: Générer des statistiques d’alignement
- Outil de statistiques Samtools avec les paramètres suivants:
- param-file « fichier BAM »:
outFile(sortie de l’outil MarkDuplicates)- « Définir la distribution de couverture »:
No- « Sortie »:
No- « Sortie »:
One single summary file- « Filtrer par drapeaux SAM »:
Do not filter- « Utiliser une séquence de référence »:
No- « Filtrer par régions »:
No
Réaligner les lectures avec lofreq viterbi
L’outil Realign reads corrige les désalignements autour des insertions et des suppressions. Ceci est nécessaire pour détecter avec précision les variantes.
hands_on Hands-on: Réaligner les lectures autour des indels
- Réaligner les lectures avec l’outil lofreq avec les paramètres suivants:
- param-file « Lit pour réaligner »:
outFile(sortie de l’outil MarkDuplicates)- » Choisissez la source du génome de référence « :
History
- fichier param « Référence »:
output(Jeu de données d’entrée)- Dans « Options avancées »:
- « Comment gérer les qualités de base de 2? »:
Keep unchanged
Ajoutez des qualités indel avec lofreq Insérez des qualités indel
Cette étape ajoute des qualités indel dans notre fichier d’alignement. Ceci est nécessaire pour appeler des variantes à l’aide de variantes d’appel avec l’outil lofreq
hands_on Hands-on: Ajouter des qualités indel
- Insérer des qualités indel avec l’outil lofreq avec les paramètres suivants :
- param-file « Reads »:
realigned(sortie de l’outil Realign reads)- « Approche de calcul Indel »:
Dindel
- « Choisissez la source du génome de référence » Dans ce cas, le fichier param « Référence » est le fichier param « Référence »:
output(Jeu de données d’entrée)
Variantes d’appel en utilisant des variantes d’appel lofreq
, nous sommes maintenant prêts à appeler des variantes.
hands_on Hands-on: Variantes d’appel
- Variantes d’appel avec l’outil lofreq avec les paramètres suivants:
- fichier param « Lecture d’entrée au format BAM »:
output(sortie de l’outil Insert indel qualities)- « Choisissez la source pour le génome de référence « :
History
- fichier param « Référence »:
output(Jeu de données d’entrée)- « Appeler des variantes à travers »:
Whole reference- « Types de variantes à appeler »:
SNVs and indels- « Paramètres d’appel de variantes »:
Configure settings
- Dans « Couverture »:
- « Couverture minimale »:
50- Dans « Appel de base »:
- « Base minimale »:
30- « Base minimale pour les bases alternatives »:
30- dans « mapping qualityy
20- « paramètres de filtre de variante »:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
La sortie de cette étape est une collection de fichiers VCF qui peuvent être visualisés dans un navigateur génomique.
Annoter les effets des variantes avec SnpEff eff:
Nous allons maintenant annoter les variantes que nous avons appelées à l’étape précédente avec l’effet qu’elles ont sur le génome du SARS-CoV-2.
hands_on Hands-on: Annoter les effets de variantes
- SnpEff eff: outil avec les paramètres suivants:
- param-file « Changements de séquence (SNPs, MNPs, InDels) »:
variants(sortie de l’outil Variantes d’appel)- « Format de sortie »:
VCF (only if input is VCF)- « Créer un rapport CSV, utile pour l’analyse en aval (-csvStats) »:
Yes- « Options d’annotation »:`
- « Sortie du filtre »:`
- « Filtrer les effets spécifiques »:
No
La sortie de cette étape est un fichier VCF avec des effets de variante ajoutés.
Créer une table de variantes à l’aide des champs d’extraction SnpSift
Nous allons maintenant sélectionner divers effets du VCF et créer un fichier tabulaire plus facile à comprendre pour les humains.
hands_on Hands-on: Créer une table de variantes
- Outil d’extraction de champs SnpSift avec les paramètres suivants:
- param-file « Fichier d’entrée de variante au format VCF »:
snpeff_output(sortie de l’outil SnpEff eff:)- « Champs à extraire »:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- « séparateur de champs multiples »:
,- « texte de champ vide »:
.
Nous pouvons inspecter les fichiers de sortie et vérifier si les variantes de ce fichier sont également décrites dans un bloc-notes observable qui montre la distribution géographique des séquences de variantes du SARS-CoV-2
Des variantes intéressantes incluent la variante C à T à la position 14408 (14408C/T) dans SRR11772204, 28144T/C dans SRR11597145 et 25563G/T dans SRR11667145.
Résumer les données avec MultiQC
Nous allons maintenant résumer notre analyse avec MultiQC, ce qui génère un beau rapport pour nos données.
hands_on Hands-on: Résumez les données
- Outil MultiQC avec les paramètres suivants:
- Dans « Résultats »:
- param – répétez « Insérer des résultats »
- « Quel outil a été utilisé pour générer des journaux? »:
fastp
- fichier param « Sortie de fastp »:
report_json(sortie de l’outil fastp)- param – répétez « Insérer des résultats »
- « Quel outil a été utilisé pour générer des journaux? »:
Samtools
- Dans « Sortie Samtools »:
- param – répétez « Insérer la sortie Samtools »
- « Type de sortie Samtools? »:
stats
- fichier param « Sortie des statistiques Samtools »:
output(sortie de l’outil de statistiques Samtools)- param-repeat « Insérer des résultats »
- « Quel outil a été utilisé pour générer des journaux ? »:
Picard
- Dans « Sortie Picard »:
- param – répétez « Insérer une sortie Picard »
- « Type de sortie Picard? »:
Markdups- fichier param « sortie Picard »:
metrics_file(sortie de l’outil MarkDuplicates)- param- répétez « Insérer des résultats »
- « Quel outil a été utilisé pour générer des journaux? »:
SnpEff
- fichier param « Sortie de SnpEff »:
csvFile(sortie de SnpEff eff:tool)
Conclusion
Félicitations, vous savez maintenant comment importer des données de séquence à partir du SRA et comment exécuter un exemple d’analyse sur ces ensembles de données.
points clés Points clés
Les données de séquence dans le SRA peuvent être directement importées dans Galaxy
Foire aux questions
Vous avez des questions sur ce tutoriel? Consultez la page FAQ pour la rubrique Analyse des variantes pour voir si votre question y figure. Si ce n’est pas le cas, veuillez poser votre question sur la chaîne GTN Gitter ou sur le Forum d’aide Galaxy
Littérature utile
De plus amples informations, y compris des liens vers la documentation et les publications originales, concernant les outils, les techniques d’analyse et l’interprétation des résultats décrits dans ce tutoriel peuvent être trouvées ici.
Feedback
Avez-vous utilisé ce matériel en tant qu’instructeur? N’hésitez pas à nous faire part de vos commentaires sur la façon dont cela s’est passé.
Citant ce tutoriel
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 De l’Archive de lecture de séquences de NCBI (SRA) à Galaxy: Analyse des variantes SARS-CoV-2 (Matériaux de formation Galaxy). / matériel de formation / sujets / analyse des variantes / tutoriels / sars-cov-2 / tutoriel.html en ligne; consulté AUJOURD’HUI
- Batut et al., 2018 Formation à l’Analyse de Données Pilotée par la Communauté pour les Systèmes Cellulaires de Biologie 10.1016 / j.cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}