Aminosäuren, Nukleotide oder irgendein anderer evolutionärer Charakter werden durch andere ersetzt. Stellen Sie sich zum Beispiel eine evolutionäre Sequenz mit drei möglichen Zuständen vor: A, B und C. Wenn das Substitutionsmodell zeitumkehrbar ist, gibt es drei Übergangsraten: A<>B, B<>C und A<>C.
Angenommen, die Raten sind 1, 1 und 0 in Einheiten der Substitution pro 100zeichen pro Zeiteinheit. Nach einer Zeiteinheit erwarten wir in einer 300 Zeichen langen Folge, die ursprünglich zu gleichen Teilen aus As, Bs und Cs besteht, eine A-B-Substitution und eine B-C-Substitution. Wenn wir zwei homologe Sequenzen in lebenden Organismen vergleichen, weil eine Zeiteinheit für beide Sequenzen vergangen ist, würden wir zwei A bis B und zwei B bis Csubstitutionen zwischen den heutigen Sequenzen erwarten.
Egal wie lange wir diesen Prozess ausführen, es wird niemals eine direkte Ersetzung von A durch C geben. Es wird auch niemals eine Ersetzung von A nach C unter einem so genannten Infinite Sites-Modell geben, bei dem nicht mehr als eine Ersetzung an einer einzelnen Site auftreten kann.Da jedoch A-zu-B- und B-zu-C-Substitutionen üblich sind, wird unter einem endlichen Sitesmodel schließlich B durch C an einer Stelle ersetzt, an der A zuvor durch B ersetzt wurde. Dieser indirekte Ersatz von A durch C (oder äquivalent in einem zeitlich reversiblen Modell, C durch A) wird wahrscheinlicher, je länger die Zeitspanne ist, in der die homologen Sequenzen getrennt werden.
Ich habe die Sequenzentwicklung basierend auf dem obigen Szenario simuliert und die Simulation für 10 Zeiteinheiten ausgeführt. Aus dieser Substitution habe ich die folgenden Zählungen für jedes Site-Muster beobachtet:
| A | B | C | |
|---|---|---|---|
| A | 91 | 9 | 0 |
| B | 5 | 86 | 9 |
| C | 0 | 9 | 91 |
Innerhalb dieser relativ kurzen es scheint jedoch nicht so, als ob eine<>Csubstitutionen aufgetreten sind. Wenn ich jedoch die Simulation für 100 Einheiten der Zeit erneut durchführe:
| A | B | C | |
|---|---|---|---|
| A | 55 | 35 | 10 |
| B | 29 | 36 | 35 |
| C | 20 | 36 | 44 |
Wie Sie sehen können, wurden viele „A“ -Zeichen durch „C“ und umgekehrt ersetzt. Allgemeiner, Unter einem Finite-Sites-Modell verursachen mehrere Substitutionen, dass die Verteilung der Site-Pattern-Zählungen viel flacher wird, als nur den Anteil der Off-Diagonalen relativ zu diagonalen Zählungen zu erhöhen.Die PAM- und BLOSUM-Score-Matrizen berücksichtigen mehrere Substitutionen auf radikal unterschiedliche Weise.
Die PAM-Matrizen für Aminosäuren wurden zusammen mit den einbuchstabigen Abkürzungen für genetisch kodierte Aminosäuren von Margaret Kirchhoff entwickelt. Sie wurden ursprünglich 1978 veröffentlicht und basieren auf den Proteinsequenzen, die Dayhoff seit den 1960er Jahren als theAtlas of Protein Sequence and Structure zusammengestellt hatte.
Der Name PAM kommt von „Point of Mutation“ und bezieht sich auf dieplazierung einer einzelnen Aminosäure in einem Protein mit einer anderen Aminosäure.Diese Mutationen wurden durch den Vergleich sehr ähnlicher Sequenzen mit mindestens 85% Identität identifiziert, und es wird angenommen, dass alle beobachteten Substitutionen das Ergebnis einer einzigen Mutation zwischen der Ahnensequenz und einer der gegenwärtigen Tagessequenzen waren.
PAM definiert auch eine Zeiteinheit, wobei 1 PAM die Zeit ist, in der erwartet wird, dass 1/100 Aminosäuren eine Mutation erfahren. Die PAM1-Wahrscheinlichkeitsmatrix zeigt die Wahrscheinlichkeit, dass die Aminosäure in Spalte j durch die Aminosäure in Zeile i ersetzt wird. Wie Sie sehen können, sind die Off-diagonalen Wahrscheinlichkeiten in der PAM1-Matrix alle sehr klein (alle Elemente wurden um 10.000 für die Lesbarkeit skaliert):
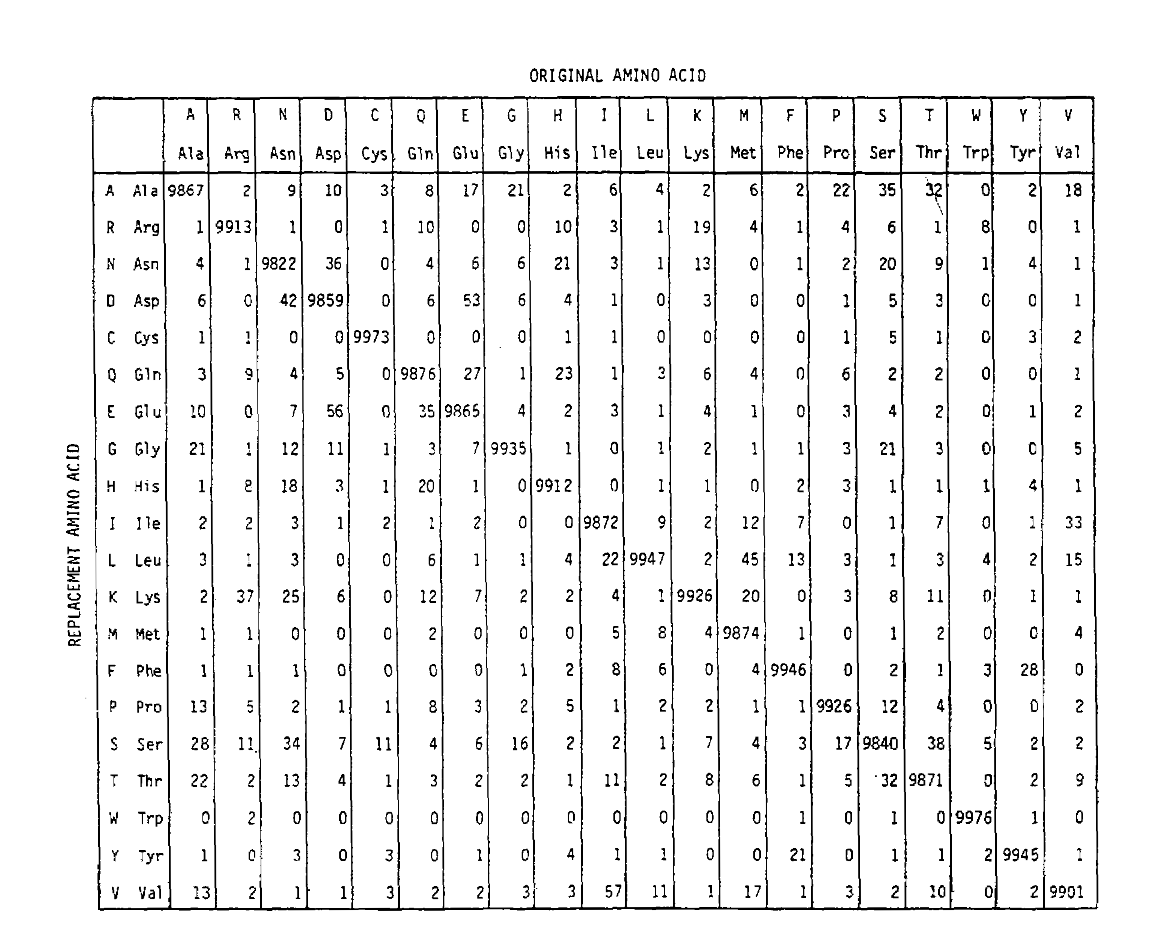
Um die Aminosäurenersatzwahrscheinlichkeiten für längere Zeitdauern zu berechnen, kann die Matrix mit der entsprechenden Anzahl von Malen multipliziert werden. So wurde die PAM250-Wahrscheinlichkeitsmatrix, die die Platzierungswahrscheinlichkeiten bei 250 PAM-Zeiteinheiten beschreibt, abgeleitet, indem die PAM1-Wahrscheinlichkeitsmatrix auf die Potenz 250 angehoben wurde (alle Elemente wurden für die Lesbarkeit um 100 skaliert):
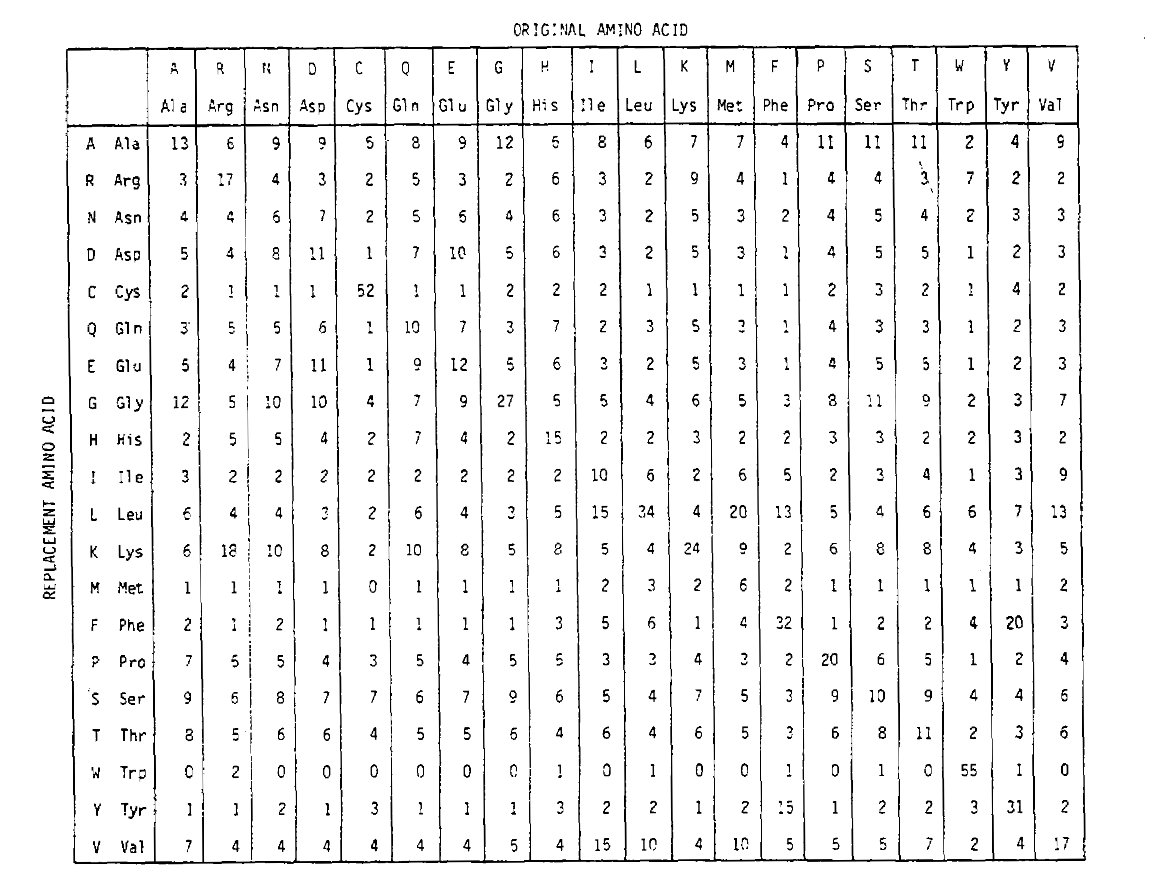
Die mit dieser Potenzierung abgeleiteten Ersetzungswahrscheinlichkeiten berücksichtigen korrekt mehrere Ersetzungen. Die Off-Diagonal-Wahrscheinlichkeiten sind nicht nur proportional größer, als Sie es für eine längere Zeitdauer erwarten würden, sondern sie sind auch flacher. Zum Beispiel ist die Wahrscheinlichkeit eines Valin (V) zu Isoleucin (I) -Ersatzes 33× größer als ein V zu histadine (H) -Ersatz in der PAM1-Matrix, aber nur 4,5× größer in der PAM250-Matrix.
Score-Matrizen können dann aus den Wahrscheinlichkeitsmatrizen und beobachteten Basisfrequenzen berechnet werden.Die BLOSUM-Matrizen, die von Steven und Jorja Henikoff entwickelt und 1992 veröffentlicht wurden, verfolgen einen ganz anderen Ansatz. Während PAM implizit ein stationäres Finite-Elemente-Modell der Evolution unter Verwendung von Matrixpotentiation anwendet, wird die Wirkung mehrerer Substitutionen implizit in BLOSUM behandelt, indem verschiedene Score-Matrizen für verschiedene Zeitskalen konstruiert werden.
Innerhalb mehrerer Sequenzausrichtungen homologer Sequenzen werden konservierte zusammenhängende Blöcke von Aminosäuren identifiziert. Innerhalb jedes Blocks werden Multiplesequences gruppiert, wenn ihre paarweise durchschnittliche Sequenzidentität höher als ein Schwellenwert ist. Der Schwellenwert beträgt 80% für die BLOSUM80-Matrix, 62% für BLOSUM62, 50% für BLOSUM50 usw.
Dies bedeutet, dass Blöcke für BLOSUM80 eine durchschnittliche paarweise Identität von nicht mehr als 80%, für BLOSUM62 von nicht mehr als 62% usw. haben.
Aminosäurenersatzwahrscheinlichkeiten für homologe Sequenzen werden aus paarweisen Vergleichen zwischen Clustern berechnet. Diese Wahrscheinlichkeiten werden das Ergebnis von Einzel- und Mehrfachsubstitutionen sein, wobei Mehrfachsubstitutionen in größeren evolutionären Entfernungen einen größeren Einfluss haben. Daher werden Scorematrizen, die aus paarweisen Vergleichen zwischen Clustern mit durchschnittlich größerer Entfernung generiert werden, wie die BLOSUM50-Matrix, natürlich den größeren Effekt mehrerer Substitutionen berücksichtigen.
Obwohl sie unterschiedliche Wege einschlagen, sind die endgültigen BLOSUM- und PAM-Score-Matrizen tatsächlich ziemlich ähnlich. Nach Henikoff und Henikoff sind die folgenden PAM- und BLOSUM-Matrizen vergleichbar:
| PAM | BLOSUM |
|---|---|
| PAM250 | BLOSUM45 |
| PAM160 | BLOSUM62 |
| PAM120 | BLOSUM80 |
For more information on PAM (Dayhoff) and BLOSUM matrices, see kapitel 2 derbiologischen Sequenzanalyse von Durbin et al., und Wikipedia.Update 13. Oktober 2019: Eine weitere Perspektive auf Substitutionsmatrizen finden Sie im Abschnitt „Umwege“ am Ende von Kapitel 5 der Bioinformatik-Algorithmen (2. oder 3. Auflage) von Compeau und Pevzner.