Ziel dieses Tutorials ist es, Sie in die Verarbeitung von Sequenzierungsdaten der nächsten Generation in Galaxy einzuführen. Dieses Tutorial verwendet eine COVID-19-Variante, die von Illumina Data aufgerufen wird, aber es geht nicht um Variantenaufrufe an sich.
Nach Abschluss dieses Tutorials wissen Sie:
- So finden Sie Daten in SRA und übertragen diese Informationen an Galaxy
- So führen Sie die grundlegende NGS-Datenverarbeitung in Galaxy durch.:
- Qualitätskontrolle (QC) von Illumina-Daten
- Zuordnung
- Entfernen von Duplikaten
- Variantenaufruf mit
lofreq - Variantenanmerkung
- Verwenden von Datasetsammlungen
- Importieren von Daten in Jupyter
### #>> In diesem Tutorial werden wir behandeln:>> 1. INHALTSVERZEICHNIS> {:Inhaltsverzeichnis}>{: .agenda}## Zwei Pfade durch dieses tutorialWe erstellt twoi Trajektorien, die Sie durch dieses Tutorial folgen können.1. ** Trajektorie 1 ** – Beginnen Sie mit der SRA der NCBI und suchen Sie nach verfügbaren Beitritten → Start (#the-sequence-read-archive)2. ** Trajektorie 2 ** – Umgehen Sie die SRA von NCBI und beginnen Sie direkt mit der Galaxie. → Start (#back-in-galaxy)Wir empfehlen, mit *** 2** zu beginnen.# Die Sequenz Read ArchiveDas (https://www.ncbi.nlm.nih.gov/sra) ist das primäre Archiv von *unassembled reads* für die (https://www.ncbi.nlm.nih.gov/). SRA ist ein großartiger Ort, um die Sequenzierungsdaten zu erhalten, die Publikationen und Studien zugrunde liegen.Dieses Tutorial behandelt, wie Sequenzdaten von SRA in Galaxy mit einer direkten Verbindung zwischen den beiden zu bekommen.> ### Kommentar Kommentar>> Sie werden auch hören, dass SRA als * Short Read Archive* bezeichnet wird, sein ursprünglicher Name.>{: .kommentar} ## Der Zugriff auf SRASRA kann entweder direkt über die Website oder über das Tool-Panel auf Galaxy erreicht werden.> ### comment Comment>> Die Option Tool panel für den Zugriff auf SRA existiert zunächst nur auf dem (https://usegalaxy.org/). Unterstützung für die direkte Verbindung zu SRA wird in der Version 20.05 von Galaxy note : enthalten sein.kommentar}> ### hands_on Hands-on: Entdecken Sie SRA Entrez>> 1. Gehen Sie zu Ihrer Galaxy-Instanz Ihrer Wahl, z. B. zu (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) oder einer anderen. (Dieses Tutorial verwendet usegalaxy.org ).> 1. Wenn Ihr Verlauf noch nicht leer ist, starten Sie einen neuen Verlauf (siehe (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) für weitere Informationen zu Galaxienverläufen)> 1. **Klicken Sie oben im Werkzeugfenster auf ** `Daten abrufen`.> 1. **Klicken Sie in der Liste der Tools unter `Daten abrufen` auf ** `SRA Server`.> Dies führt Sie zum (https://www.ncbi.nlm.nih.gov/sra) — Sie können auch direkt von der SRA starten. Ein Suchfeld wird oben auf der Seite angezeigt. Versuchen Sie, nach etwas zu suchen, an dem Sie interessiert sind, z. B. `Dolphin` oder `kidney` oder `Dolphin kidney`, und klicken Sie dann ** auf ** die Schaltfläche `Search`.>> Dies gibt eine Liste von *SRA-Experimenten* zurück, die Ihrer Suchzeichenfolge entsprechen. SRA-Experimente, auch bekannt als * SRX-Einträge *, enthalten Sequenzdaten aus einem bestimmten Experiment sowie eine Erklärung des Experiments selbst und anderer verwandter Daten. Sie können die zurückgegebenen Experimente erkunden, indem Sie auf ihren Namen klicken. Siehe (https://www.ncbi.nlm.nih.gov/books/NBK56913/) in der (https://www.ncbi.nlm.nih.gov/books/n/helpslib/) für mehr.>> Wenn Sie Text in das SRA-Suchfeld eingeben, verwenden Sie (https://www.ncbi.nlm.nih.gov/sra/docs/srasearch/). Entrez unterstützt sowohl einfache Textsuchen als auch sehr präzise Suchen, die bestimmte Metadaten überprüfen und beliebig komplexe logische Ausdrücke verwenden. Mit Entrez können Sie Ihre Suche von einfach bis fortgeschritten skalieren, während Sie Ihre Suche eingrenzen. Die Syntax der erweiterten Suche kann entmutigend erscheinen, aber SRA bietet eine grafische (https://www.ncbi.nlm.nih.gov/sra/advanced/), um die spezifische Syntax zu generieren. Und wie wir weiter unten sehen werden, bietet der SRA Run Selector eine noch freundlichere Benutzeroberfläche zum Eingrenzen unserer ausgewählten Daten.>> Spielen Sie mit der SRA Entrez-Oberfläche, einschließlich des erweiterten Abfrage-Builders, um zu sehen, ob Sie eine Reihe von SRA-Experimenten identifizieren können, die für einen Ihrer Forschungsbereiche relevant sind.{: .hands_on}> ### hands_on Zum Anfassen: Generieren Liste der passenden Experimente mit Entrez>> Nun, da Sie eine grundlegende Vertrautheit mit SRA Entrez haben, lassen Sie uns die Sequenzen in diesem Tutorial verwendet finden.>> 1. Wenn Sie noch nicht dort sind, **navigieren Sie** zurück zu (https://www.ncbi.nlm.nih.gov/sra> 1. ** Löschen ** jeder Suchtext aus dem Suchfeld.> 1. ** Geben Sie ** ’sars-cov-2` in das Suchfeld ein und ** klicken Sie auf ** `Suchen‘.> Dies gibt eine längere Liste von SRA-Experimenten zurück, die unserer Suche entsprechen, und diese Liste ist viel zu lang, um sie in einer Tutorial-Übung zu verwenden. An dieser Stelle könnten wir den erweiterten Entrez Query Builder verwenden, den wir oben kennengelernt haben.> Aber das werden wir nicht. Stattdessen senden wir die *zu lange für ein Tutorial * Listenergebnisse, die wir haben, an den SRA Run Selector und verwenden seine freundlichere Schnittstelle, um unsere Ergebnisse einzugrenzen.>> !(../../bilder/sra_entrez.png){: .hands_on}> ### hands_on Zum Anfassen: Wechseln Sie von Entrez zu SRA Run Selector>> Zeigen Sie die Ergebnisse mit dem RunSelector als erweiterte interaktive Tabelle an.>> 1. Klicken Sie auf Ergebnisse senden, um die Auswahl auszuführen, die in einem Feld oben in den Suchergebnissen angezeigt wird.>> !(../../bilder/sra_entrez_result.png)>>> ### Tipp Was ist, wenn Sie den Link Auswahl Ausführen nicht sehen?>>>> Möglicherweise haben Sie diesen Text früher bemerkt, als Sie die Entrez-Suche erkundeten. Dieser Text erscheint nur manchmal, wenn die Anzahl der Suchergebnisse in ein ziemlich breites Fenster fällt. Sie werden es nicht sehen, wenn Sie nur wenige Ergebnisse haben, und Sie werden es nicht sehen, wenn Sie mehr Ergebnisse haben, als der Run Selector akzeptieren kann.>>>> * Sie müssen den Selektor ausführen, um Ihre Ergebnisse an Galaxy zu senden.* Was ist, wenn Sie nicht genügend Ergebnisse haben, um die Anzeige dieses Links auszulösen? In diesem Fall rufen Sie get to the Run Selector auf, indem Sie ** auf ** im Pulldown-Menü `Senden an` oben rechts im Ergebnisfenster klicken. Um den Selektor auszuführen, ** wählen Sie ** `Selektor ausführen` und klicken Sie dann ** auf ** die Schaltfläche `Los‘.> !(../../bilder/sra_entrez_send_to.png)> {: .tipp}>>> 1. ** Klicken Sie oben im Suchergebnisfenster auf ** `Ergebnisse an Run Selector senden`. (Wenn Sie diesen Link nicht sehen, lesen Sie den Kommentar direkt oben.){: .hands_on}## SRA Run Selectorwir haben bereits gelernt, wie wir unsere Suchergebnisse mithilfe der erweiterten Syntax von Entrez eingrenzen können. Wir haben diese Kraft jedoch nicht genutzt, als wir in Entrez waren. Stattdessen haben wir eine einfache Suche verwendet und dann alle Ergebnisse an den Run Selector gesendet. Wir haben noch nicht die (kurze) Liste der Ergebnisse, an denen wir die Analyse durchführen möchten. *Was machen wir?* Wir verwenden Entrez und den Run Selector so, wie sie verwendet werden sollen: * Verwenden Sie die Entrez-Schnittstelle, um Ihre Ergebnisse auf eine Größe einzugrenzen, die der Run Selector verbrauchen kann. * Senden Sie diese Entrez-Ergebnisse an den SRA-Laufselektor * Verwenden Sie die viel freundlichere Benutzeroberfläche des Laufselektors, um 1. Leichter zu verstehen, die Daten, die wir haben 1. Grenzen Sie diese Ergebnisse mit diesem Wissen ein.> ### Kommentar Run Selector ist sowohl mehr als auch weniger als Entrez>> Run Selector kann das meiste, aber nicht alles, was die Entrez-Suchsyntax kann. Run Selector verwendet die * facettierte Suche * -Technologie, die einfach zu bedienen und leistungsstark ist, aber inhärente Grenzen hat. Insbesondere funktioniert Entrez besser, wenn nach Attributen gesucht wird, die Dutzende, Hunderte oder Tausende verschiedener Werte haben. Run Selector funktioniert besser bei der Suche nach Attributen mit weniger als 20 verschiedenen Werten. Glücklicherweise beschreibt das die meisten Suchanfragen.{: .kommentar}Das Auswahlfenster Ausführen ist in mehrere Bereiche unterteilt: * ** `Filterliste’**: In der oberen linken Ecke. Hier werden wir unsere Suche verfeinern.* ** ‚Select‘ **: Eine Zusammenfassung dessen, was ursprünglich an Run Selector übergeben wurde und wie viel davon wir bisher ausgewählt haben. (Und bisher haben wir nichts davon ausgewählt.) Beachten Sie auch die verlockende, aber immer noch ausgegraut, `Galaxy` Taste.* ** `Found x Items‘ ** Dies ist zunächst die Liste der Elemente, die von Entrez an Run Selector gesendet wurden. Diese Liste wird verkleinert, wenn wir Filter darauf anwenden.!(../../bilder/sra_run_selector.png)> ### Kommentar Warum ist die Anzahl der gefundenen Objekte * gestiegen?*>> Denken Sie daran, dass die Entrez-Schnittstelle SRA-Experimente (SRX-Einträge) auflistet. Run Selector listet * Runs * auf – Sequenzierungsdatensätze – und es gibt * einen oder mehrere * Runs pro Experiment. Wir haben die gleichen Daten wie zuvor, wir sehen sie jetzt nur noch detaillierter.{: .kommentar}Die ‚Filterliste‘ oben links zeigt Spalten in unseren Ergebnissen an, die entweder fortlaufende numerische Werte oder 10 oder weniger (Sie können diese Zahl ändern) unterschiedliche Werte enthalten. ** Blättern ** nach unten durch die Liste wählen Sie einige der Filter. Wenn ein Filter ausgewählt ist, wird unten ein Feld *Werte * angezeigt, in dem die Optionen für diesen Filter und die Anzahl der Durchläufe mit jeder Option aufgeführt sind. Diese Werte / Optionen werden aus den Dataset-Metadaten abgerufen. Versuchen Sie ** Auswahl ** ein paar interessant klingende Filter und dann ** wählen ** eine oder mehrere Optionen für jeden Filter. Versuchen Sie ** Deaktivieren ** Optionen und Filter. Während Sie dies tun, verringert oder erhöht sich die Anzahl der gefundenen Ergebnisse.> ### Tipp Tipp: Verwenden Sie Filter, um die Daten besser zu verstehen>> Mit Filtern können Sie die betrachteten Datensätze für das Senden an Galaxy eingrenzen, aber sie sind auch eine hervorragende Möglichkeit, Ihre Daten zu verstehen:> Zunächst ist die Auswahl eines Filters eine einfache Möglichkeit, den Wertebereich in einer Spalte anzuzeigen. Sie können möglicherweise nicht (https://www.google.com/search?q=sra+sirs_outcome), aber Sie können es möglicherweise herausfinden, indem Sie sehen, welche Werte darin enthalten sind.> Zweitens können Sie untersuchen, wie sich verschiedene Spalten aufeinander beziehen. Gibt es eine Beziehung zwischen `sirs_outcome`-Werten und `disease_stage`-Werten?{: .tipp}> ### hands_on Hands-on: Verfeinern Sie Ihre Ergebnisse mit dem Run Selector>> 1. Wenn Sie Filter aktiviert haben, ** deaktivieren ** sie.> Sobald Sie dies getan haben, werden unter der „Filterliste“ keine * Werte * -Felder mehr angezeigt.> 2. ** Kopieren und einfügen ** Diese Suchzeichenfolge in das Suchfeld `Gefundene Objekte‘.>> SRR11772204 ODER SRR11597145 ODER SRR11667145>> Dieser handverlesene Satz von Läufen begrenzt unsere Ergebnisse zu 3 läuft aus verschiedenen geographischen verteilung.{: .hands_on} Dies reduziert Ihre Liste der gefundenen Elemente von Zehntausenden von Läufen auf 3 Läufe (eine überschaubare Anzahl für ein Tutorial!). Aber wir sind noch nicht ganz fertig mit Run Selector. Beachten Sie, dass die Schaltfläche `Galaxy` immer noch ausgegraut ist. Wir haben unsere Optionen eingegrenzt, aber wir haben noch nichts ausgewählt, was an Galaxy gesendet werden soll.Sie können jeden verbleibenden Lauf auswählen, indem Sie ** auf ** das Häkchen oben in der ersten Spalte klicken. Sie können alles deaktivieren, indem Sie ** auf ** das `X‘ klicken.> ### hands_on Hands-on: Läufe auswählen und an Galaxy senden>> 1. Wählen Sie alle Läufe aus, indem Sie ** auf ** das `X‘ klicken.> Und jetzt ist der „Galaxy“ -Button live.> 1. ** Klicken Sie oben auf der Seite auf die Schaltfläche `Galaxy` im Abschnitt `Select‘.{: .hands_on} ## Zurück in GalaxyWenn wir im Run Selector auf `Galaxy` klicken, passieren mehrere Dinge. Zuerst wird ein neuer Browser-Tab oder ein neues Fenster gestartet, das in Galaxy geöffnet wird. Sie sehen das * große grüne Kästchen *, das anzeigt, dass der Handshake zwischen SRA und Galaxy erfolgreich war, und Sie sehen dann einen neuen SRA-Job in Ihrem Verlaufsfenster. Dieses Feld kann als grau / ausstehend beginnen, was darauf hinweist, dass die Übertragung noch nicht begonnen hat, oder es kann direkt zu gelb / Laufen oder zu grün / fertig gehen.> ### hands_on Zum Anfassen: Untersuchen Sie den neuen SRA-Datensatz>> 1. Sobald die `SRA‘-Übertragung abgeschlossen ist, ** klicken Sie ** auf das Galaxy-Eye-Symbol (Auge) des Datensatzes.>> Dies zeigt den Datensatz im mittleren Bereich von Galaxy an.{: .hands_on} Das `SRA‘-Dataset sind keine Sequenzdaten, sondern * Metadaten *, die wir verwenden, um Sequenzdaten von SRA abzurufen. Diese Metadaten spiegeln die Informationen wider, die wir im Abschnitt `Gefundene Objekte` des Laufselektors gesehen haben. Die Metadaten sind nicht die Enddaten, die wir von SRA suchen, aber all diese Metadaten sind oft in nachfolgenden Analyseschritten nützlich.Verwenden wir nun diese Metadaten, um die Sequenzdaten von SRA abzurufen. SRA bietet Werkzeuge zum Extrahieren aller Arten von Informationen, einschließlich der Sequenzdaten selbst. Das Galaxy-Tool „Faster Download and Extract Reads in FASTQ“ basiert auf dem Dienstprogramm SRA (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump) und macht genau das.–>
- Finden Sie die erforderlichen Daten in SRA
- hands_on Hands-on: Aufgabenbeschreibung
- Kommentar Kommentar
- Prozess und Filter SraRunInfo.csv-Datei in der Galaxie
- hands_on Hands-on: SraRunInfo hochladen.csv-Datei in Galaxy
- Kommentar Vorsicht vor Schnitten
- hands_on Hands-on: Erstellen einer Teilmenge von Daten
- Tipp Tipp: Werkzeuge finden
- Laden Sie Sequenzierungsdaten mit schnellerem Download herunter und extrahieren Sie Lesevorgänge in FASTQ
- hands_on Hands-on: Aufgabenbeschreibung
- Was nun?
- Variationsanalyse von SARS-Cov-2-Sequenzierungsdaten
- kommentieren Sie Die usegalaxy.* COVID-19-Analyseprojekt
- Holen Sie sich die Referenzgenomdaten
- hands_on Hands-on: Holen Sie sich die Referenzgenomdaten
- Tipp: Importieren über Links
- Adaptertrimmen mit fastp
- hands_on Hands-on: Aufgabenbeschreibung
- Ausrichtung mit Karte mit BWA-MEM
- hands_on Zum Anfassen: Richten Sie Sequenzierungslesungen an Referenzgenom aus
- Duplikate mit dem Werkzeug MarkDuplicates entfernen
- hands_on Hands-on: PCR-Duplikate entfernen
- Alignment-Statistiken mit Samtools stats generieren
- hands_on Zum Anfassen: Ausrichtungsstatistik erzeugen
- Neuausrichtung liest mit lofreq viterbi
- hands_on Hands-on: Reads um Indels neu ausrichten
- Indel-Qualitäten mit lofreq hinzufügen Indel-Qualitäten einfügen
- hands_on Hands-on: Fügen Sie Indel-Qualitäten hinzu
- Aufrufvarianten mit lofreq Aufrufvarianten
- hands_on Hands-on: Varianten aufrufen
- Varianteneffekte mit SnpEff annotieren eff:
- hands_on Hands-on: Varianteneffekte annotieren
- Variantentabelle mit SnpSift-Extraktfeldern erstellen
- hands_on Hands-on: Variantentabelle erstellen
- Daten mit MultiQC zusammenfassen
- hands_on Hands-on: Daten zusammenfassen
- Fazit
- Schlüsselpunkte Schlüsselpunkte
- Häufig gestellte Fragen
- Nützliche Literatur
- Feedback
- Zitieren Sie dieses Tutorial
- details BibTeX
Finden Sie die erforderlichen Daten in SRA
Zuerst müssen wir einen guten Datensatz zum Spielen finden. Das Sequence Read Archive (SRA) ist das primäre Archiv von Unassembled Reads, das von den US National Institutes of Health (NIH) betrieben wird. SRA ist ein großartiger Ort, um die Sequenzierungsdaten zu erhalten, die Publikationen und Studien zugrunde liegen. Machen wir das:
hands_on Hands-on: Aufgabenbeschreibung
- Gehen Sie zur SRA-Seite von NCBI, indem Sie Ihren Browser auf https://www.ncbi.nlm.nih.gov/sra
- Geben Sie im Suchfeld
SARS-CoV-2 Patient Sequencing From Partners / MGH(Alternativ klicken Sie einfach auf diesen Link)
- Die Webseite zeigt eine große Anzahl von SRA-Datensätzen (zum Zeitpunkt des Schreibens gab es 2.223). Dies sind Daten aus einer Studie, die die Analyse von SARS-CoV-2 in der Region Boston beschreibt.
- Laden Sie Metadaten herunter, die diese Datensätze beschreiben:
- Klicken Sie auf Senden an: Dropdown
- Auswählen von
File- Ändern des Formats in
RunInfo- Klicken Datei erstellenhier sollte es aussehen:
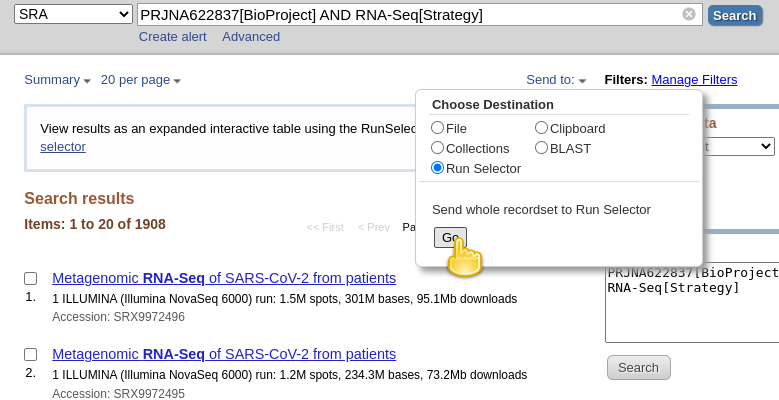
- Dies würde eine ziemlich große
SraRunInfo.csvDatei in IhremDownloadsOrdner erstellen.
Nachdem wir diese Datei heruntergeladen haben, können wir zu einer Galaxy-Instanz gehen und mit der Verarbeitung beginnen.
Kommentar Kommentar
Beachten Sie, dass die Datei, die wir gerade heruntergeladen haben, keine Sequenzdaten selbst ist. Vielmehr sind es Metadaten, die Eigenschaften von Sequenzierungslesungen beschreiben. Wir werden diese Liste auf nur wenige Beitritte filtern, die im Rest dieses Tutorials verwendet werden.
Prozess und Filter SraRunInfo.csv-Datei in der Galaxie
hands_on Hands-on: SraRunInfo hochladen.csv-Datei in Galaxy
- Gehen Sie zu Ihrer Galaxy-Instanz Ihrer Wahl, z usegalaxy.org, usegalaxy.eu, usegalaxy.org.au oder irgendein anderes. (Dieses Tutorial verwendet usegalaxy.org ).
- Klicken Sie auf die Schaltfläche Daten hochladen:
- Klicken Sie im angezeigten Dialogfeld auf die Schaltfläche „Lokale Dateien auswählen“:

- Suchen und wählen Sie
SraRunInfo.csvDatei von Ihrem Computer- Klicken Sie auf die Schaltfläche Start
- Sie können nun den Inhalt dieser Datei anzeigen, indem Sie auf galaxy-Auge (Auge) Symbol. Sie werden sehen, dass diese Datei viele Informationen zu einzelnen SRA-Beitritten enthält. In dieser Studie entspricht jeder Beitritt einem einzelnen Patienten, dessen Proben sequenziert wurden.
Galaxy kann alle 2.000+ Datensätze verarbeiten, aber um dieses Tutorial erträglich zu machen, müssen wir eine kleinere Teilmenge auswählen. Insbesondere unsere bisherigen Erfahrungen mit diesen Daten zeigen zwei interessante Datensätze SRR11954102 und SRR12733957. Also, lassen Sie uns sie herausziehen.
Kommentar Vorsicht vor Schnitten
Der praktische Abschnitt unten verwendet das Schnittwerkzeug. Es gibt zwei Schnittwerkzeuge in Galaxy aus historischen Gründen. In diesem Beispiel wird das Werkzeug mit dem vollständigen Namen Spalten aus einer Tabelle ausschneiden (ausschneiden) verwendet. Die gleiche Logik gilt jedoch für das andere Tool. Es hat einfach eine etwas andere Schnittstelle.
hands_on Hands-on: Erstellen einer Teilmenge von Daten
- Werkzeug „Zeilen auswählen, die einem Ausdruck entsprechen“ im Abschnitt „Filtern und Sortieren“ des Werkzeugfensters finden.
Tipp Tipp: Werkzeuge finden
Auf Galaxy ist möglicherweise eine überwältigende Anzahl von Werkzeugen installiert. Um ein bestimmtes Werkzeug zu finden, geben Sie den Werkzeugnamen in das Suchfeld des Werkzeugbedienfelds ein, um das Werkzeug zu finden.
- Stellen Sie sicher, dass der
SraRunInfo.csvDatensatz, den wir gerade hochgeladen haben, in der param-Datei „Select lines from“ des Werkzeugformulars aufgeführt ist.- Geben Sie im Feld „das Muster“ den folgenden Ausdruck ein →
SRR12733957|SRR11954102. Dies sind zwei Zeichen, die wir durch das Pipe-Symbol getrennt finden möchten|. Das|bedeutetor: Suchen Sie Zeilen, dieSRR12733957oderSRR11954102.- Klicken Sie auf
ExecuteTaste.- Dadurch wird eine Datei generiert, die zwei Zeilen enthält (nun … eine Zeile wird auch als Header verwendet, sodass angezeigt wird, dass die Datei drei Zeilen enthält. Es ist OK.)
- Schneiden Sie die erste Spalte aus der Datei mit Werkzeug „Cut“ Werkzeug, das Sie in Textmanipulation Abschnitt des Werkzeugfensters finden.
- Stellen Sie sicher, dass der im vorherigen Schritt erstellte Datensatz im Feld „Zu schneidende Datei“ des Werkzeugformulars ausgewählt ist.
- Ändern Sie „Begrenzt durch“ in
Comma- Wählen Sie in „Liste der Felder“
Column: 1.- Hit
ExecuteDies wird eine Textdatei mit nur zwei Zeilen erzeugen:SRR12733957SRR11954102
Nun, da wir Bezeichner von Datensätzen haben, die wir wollen, müssen wir die tatsächlichen Sequenzierungsdaten herunterladen.
Laden Sie Sequenzierungsdaten mit schnellerem Download herunter und extrahieren Sie Lesevorgänge in FASTQ
hands_on Hands-on: Aufgabenbeschreibung
- Schnelleres Herunterladen und Extrahieren von Lesevorgängen in FASTQ Tool mit den folgenden Parametern:
- „Eingabetyp auswählen“:
List of SRA accession, one per line
- Der Parameter param-file „sra-list“ sollte die Ausgabe des Werkzeugs „Cut“ aus dem vorherigen Schritt zeigen.
- Klicken Sie auf die Schaltfläche
Execute. Dadurch wird das Tool ausgeführt, das die Sequenzlesedatensätze für die Läufe abruft, die imSRA-Datensatz aufgelistet wurden. Es kann einige Zeit dauern. Dies kann also ein guter Zeitpunkt sein, um Kaffee zu holen.- Mehrere Einträge werden in Ihrem History-Panel erstellt, wenn Sie diesen Job einreichen:
Pair-end data (fasterq-dump): Enthält Gepaarte Datensätze (falls verfügbar)Single-end data (fasterq-dump)Enthält Single-End-Datensätze (falls verfügbar)Other data (fasterq-dump)Enthält Ungepaarte Datensätze (falls verfügbar)fasterq-dump logEnthält Informationen zur Werkzeugausführung
Die ersten drei Elemente sind eigentlich Sammlungen von Datensätzen. Sammlungen in Galaxy sind logische Gruppierungen von Datensätzen, die die semantischen Beziehungen zwischen ihnen im Experiment / in der Analyse widerspiegeln. In diesem Fall erstellt das Tool jeweils eine separate Sammlung für gepaarte Lesevorgänge, einzelne Lesevorgänge und andere.Weitere Informationen finden Sie in den Collections-Tutorials.
Erkunden Sie die Sammlungen, indem Sie zuerst auf den Namen der Sammlung im Verlaufsfenster klicken. Dies führt Sie in die Sammlung und zeigt Ihnen die darin enthaltenen Datensätze. Sie können dann zurück zur äußeren Ebene Ihres Verlaufs navigieren.
Sobald fasterq die Datenübertragung abgeschlossen hat (alle Felder sind grün / fertig), können wir sie analysieren.
Was nun?
Sie können die abgerufenen Daten jetzt mit beliebigen Sequenzanalysetools und Workflows in Galaxy analysieren. SRA hält Backing-Daten für jede erdenkliche Art von *-seq Experiment.
Wenn Sie dieses Tutorial ausgeführt haben, aber Datensätze abgerufen haben, an denen Sie interessiert waren, finden Sie im Rest der GTN-Bibliothek Ideen zur Analyse in Galaxy.
Wenn Sie jedoch die in den obigen Beispielen dieses Tutorials verwendeten Datensätze abgerufen haben, können Sie die folgende SARS-CoV-2-Variantenanalyse ausführen.
Variationsanalyse von SARS-Cov-2-Sequenzierungsdaten
In diesem Teil des Tutorials führen wir Variantenaufrufe und grundlegende Analysen der oben heruntergeladenen Datensätze durch. Wir beginnen mit dem Herunterladen der Wuhan-Hu-1 SARS-CoV-2-Referenzsequenz, führen dann Adaptertrimmen, Ausrichten und Variantenaufruf aus und betrachten schließlich die geografische Verteilung einiger der gefundenen Varianten.
kommentieren Sie Die usegalaxy.* COVID-19-Analyseprojekt
Dieses Tutorial verwendet eine Teilmenge der Daten und durchläuft den Abschnitt Variationsanalyse von covid19.galaxyprojekt.Organisierungstafel.Die Daten für covid19.galaxyproject.org wird laufend aktualisiert, sobald neue Datensätze veröffentlicht werden.
Holen Sie sich die Referenzgenomdaten
Die Referenzgenomdaten für heute beziehen sich auf SARS-CoV-2, „Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome“, mit der Accession ID von NC_045512.2.
Diese Daten sind bei Zenodo unter folgendem Link verfügbar.
hands_on Hands-on: Holen Sie sich die Referenzgenomdaten
Importieren Sie die folgende Datei in Ihre Historie:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzTipp: Importieren über Links
- Kopieren Sie den Link-Speicherort
- Öffnen Sie den Galaxy Upload Manager (galaxy-upload oben rechts im Werkzeugfenster)
- Wählen Sie Einfügen/Daten abrufen
- Fügen Sie den Link in das Textfeld ein
- Drücken Sie Start
- Schließen Sie das Fenster Standardmäßig verwendet Galaxy die URL als Namen.
Adaptertrimmen mit fastp
Das Entfernen von Sequenzierungsadaptern verbessert Alignments und Variantenaufrufe. das fastp-Tool kann weit verbreitete Sequenzierungsadapter automatisch erkennen.
hands_on Hands-on: Aufgabenbeschreibung
- fastp-Tool mit den folgenden Parametern:
- „Single-End oder Gepaarte Sammlung“:
Paired Collection
- param-file „Gepaarte Sammlung(en) auswählen“:
list_paired(Ausgabe von schnelleren Download- und Extraktlesungen im FASTQ-Tool)- In „Ausgabeoptionen“:
- „JSON-Bericht ausgeben“:
Yes
Ausrichtung mit Karte mit BWA-MEM
Das BWA-MEM-Tool ist ein weit verbreiteter Sequenzausrichter für Sequenzierungsdatensätze mit kurzer Lesezeit, wie wir sie in diesem Lernprogramm analysieren.
hands_on Zum Anfassen: Richten Sie Sequenzierungslesungen an Referenzgenom aus
- Karte mit dem BWA-MEM-Tool mit den folgenden Parametern:
- „Wählen Sie ein Referenzgenom aus Ihrer Historie aus oder verwenden Sie einen integrierten Index?“:
Use a genome from history and build index
- param-file „Verwenden Sie den folgenden Datensatz als Referenzsequenz“:
output(Eingabedatensatz)- „Single- oder Paired-End-Lesevorgänge“:
Paired Collection
- param-file “ collection“:
output_paired_coll(Ausgabe des Fastp-Tools)- „Informationen zum Lesen von Gruppen festlegen?“:
Do not set- „Analysemodus auswählen“:
1.Simple Illumina mode
Duplikate mit dem Werkzeug MarkDuplicates entfernen
Das Werkzeug MarkDuplicates entfernt doppelte Sequenzen, die aus Artefakten zur sequenzierung von Artefakten. Es ist wichtig, diese Artefaktsequenzen zu entfernen, um eine künstliche Überrepräsentation einzelner Moleküle zu vermeiden.
hands_on Hands-on: PCR-Duplikate entfernen
- Markierenduplikate Werkzeug mit den folgenden Parametern:
- param-file „SAM/BAM-Datensatz oder Datensatzsammlung auswählen“:
bam_output(Ausgabe der Karte mit dem BWA-MEM-Tool)- „Wenn true, schreiben Sie keine Duplikate in die Ausgabedatei, anstatt sie mit den entsprechenden gesetzten Flags zu schreiben“:
Yes
Alignment-Statistiken mit Samtools stats generieren
Nach dem obigen Schritt der doppelten Markierung können wir Statistiken über die von uns generierte Ausrichtung generieren.
hands_on Zum Anfassen: Ausrichtungsstatistik erzeugen
- Samtools stats tool mit folgenden Parametern:
- param-file „BAM file“:
outFile(Ausgabe von MarkDuplicates tool)- „Set coverage distribution“:
No- „Output“:
One single summary file- „Nach SAM-Flags filtern“:
Do not filter- „Referenzsequenz verwenden“:
No- „Nach Regionen filtern“:
No
Neuausrichtung liest mit lofreq viterbi
Neuausrichtung liest Werkzeug korrigiert Fehlausrichtungen um Einfügungen und Löschungen. Dies ist erforderlich, um Varianten genau zu erkennen.
hands_on Hands-on: Reads um Indels neu ausrichten
- Reads mit lofreq tool mit folgenden Parametern neu ausrichten:
- param-file „Reads to realign“:
outFile(Ausgabe von MarkDuplicates tool)- „Wählen Sie die das Referenzgenom“:
History
- param-Datei „Referenz“:
output(Eingabedatensatz)- In „Erweiterte Optionen“:
- „Wie gehe ich mit Basisqualitäten von 2 um?“:
Keep unchanged
Indel-Qualitäten mit lofreq hinzufügen Indel-Qualitäten einfügen
Dieser Schritt fügt Indel-Qualitäten in unsere Alignment-Datei ein. Dies ist notwendig, um Varianten mit dem lofreq-Tool aufzurufen
hands_on Hands-on: Fügen Sie Indel-Qualitäten hinzu
- Fügen Sie Indel-Qualitäten mit dem lofreq-Tool mit den folgenden Parametern ein:
- param-file „Reads“:
realigned(Ausgabe des Reads-Tools neu ausrichten)- „Indel-Berechnungsansatz“:
Dindel
- „Wählen Sie die Quelle für das Referenzgenom“:
History
- param-Datei „Referenz“:
output(Eingabedatensatz)
Aufrufvarianten mit lofreq Aufrufvarianten
Wir sind jetzt bereit, Varianten aufzurufen.
hands_on Hands-on: Varianten aufrufen
- Varianten aufrufen mit dem lofreq-Tool mit folgenden Parametern:
- param-file „Eingabe liest im BAM-Format“:
output(Ausgabe des Indelete-Tools einfügen)- „Wählen Sie die Quelle für das Referenzgenom“:
History
- param-file „Reference“:
output(Eingabedatensatz)- „Variantentypen aufrufen“:
Whole reference- „Variantentypen aufrufen“:
SNVs and indels- „Variant calling parameters“:
Configure settings
- In „Coverage“:
- „Minimal coverage“:
50- In „Base-calling“ minimum BASEQ“:
- “ Minimum BASEQ“:
30- “ Minimum BASEQ for alternate bases“:
30- In“ mapping quality gif“:
- “ minimum mapping quality^“:
20„variant filter parameters“:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
Die Ausgabe dieses Schritts ist eine Sammlung von VCF-Dateien, die in einem Genombrowser visualisiert werden können.
Varianteneffekte mit SnpEff annotieren eff:
Wir werden nun die Varianten, die wir im vorherigen Schritt aufgerufen haben, mit den Auswirkungen auf das SARS-CoV-2-Genom annotieren.
hands_on Hands-on: Varianteneffekte annotieren
- SnpEff eff: Werkzeug mit folgenden Parametern:
- param-Datei „Sequenzänderungen (SNPs, MNPs, InDels)“:
variants(Ausgabe des Callback-Tools)- „Ausgabeformat“:
VCF (only if input is VCF)- „CSV-Bericht erstellen, nützlich für die Downstream-Analyse (-csvStats)“:
Yes- „Annotationsoptionen“: `
- „Filterausgabe“ : `
- „Spezifische Effekte herausfiltern“:
No
Die Ausgabe dieses Schritts ist eine VCF-Datei mit zusätzlichen Varianteneffekten.
Variantentabelle mit SnpSift-Extraktfeldern erstellen
Wir werden nun verschiedene Effekte aus der VCF auswählen und eine tabellarische Datei erstellen, die für den Menschen leichter verständlich ist.
hands_on Hands-on: Variantentabelle erstellen
- SnpSift Werkzeug zum Extrahieren von Feldern mit folgenden Parametern:
- param-file „Varianteneingabedatei im VCF-Format“:
snpeff_output(Ausgabe von SnpEff eff: tool)- „Zu extrahierende Felder“ :
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- „Trennzeichen für mehrere Felder“:
,- „leerer Feldtext“:
.
Wir können die Ausgabedateien inspizieren und prüfen, ob Varianten in dieser Datei auch in einem beobachtbaren Notizbuch beschrieben sind, das die geografische Verteilung von SARS-Viren in CoV-2-Variantensequenzen
Interessante Varianten sind die C-T-Variante an Position 14408 (14408C / T) in SRR11772204, 28144T / C in SRR11597145 und 25563G / T in SRR11667145.
Daten mit MultiQC zusammenfassen
Wir werden nun unsere Analyse mit MultiQC zusammenfassen, wodurch ein schöner Bericht für unsere Daten erstellt wird.
hands_on Hands-on: Daten zusammenfassen
- MultiQC-Tool mit den folgenden Parametern:
- In „Ergebnisse“:
- param-repeat „Ergebnisse einfügen“
- „Welches Tool wurde verwendet Protokolle generieren?“:
fastp
- param-file „Ausgabe von fastp“:
report_json(Ausgabe des Fastp-Tools)- param-repeat „Ergebnisse einfügen“
- „Welches Tool wurde verwendet, um Protokolle zu generieren?“:
Samtools
- In „Samtools-Ausgabe“:
- param-repeat „Samtools-Ausgabe einfügen“
- „Art der Samtools-Ausgabe?“:
stats
- param-Datei „Samtools stats Ausgabe“:
output(Ausgabe von Samtools stats tool)- param-repeat „Ergebnisse einfügen“
- „Welches Tool wurde verwendet, um Protokolle zu generieren?“:
Picard
- In „Picard-Ausgabe“:
- param-repeat „Picard-Ausgabe einfügen“
- „Art der Picard-Ausgabe?“:
Markdups- param-Datei „Picard-Ausgabe“:
metrics_file(Ausgabe von MarkDuplicates tool)- param-repeat „Ergebnisse einfügen“
- „Welches Tool wurde verwendet, um Protokolle zu generieren?“:
SnpEff
- param-file „Ausgabe von SnpEff“:
csvFile(Ausgabe von SnpEff eff: tool)
Fazit
Herzlichen Glückwunsch, Sie wissen jetzt, wie Sie Sequenzdaten aus der SRA importieren und eine Beispielanalyse für diese Datensätze durchführen.
Schlüsselpunkte Schlüsselpunkte
Sequenzdaten in der SRA können direkt in Galaxy importiert werden
Häufig gestellte Fragen
Haben Sie Fragen zu diesem Tutorial? Schauen Sie sich die FAQ-Seite für das Thema Variantenanalyse an, um zu sehen, ob Ihre Frage dort aufgeführt ist. Wenn nicht, stellen Sie bitte Ihre Frage auf dem GTN Gitter Channel oder im Galaxy Help Forum
Nützliche Literatur
Weitere Informationen, einschließlich Links zu Dokumentationen und Originalpublikationen, zu den in diesem Tutorial beschriebenen Tools, Analysetechniken und der Interpretation der Ergebnisse finden Sie hier.
Feedback
Hast du dieses Material als Instruktor verwendet? Fühlen Sie sich frei, uns Feedback zu geben, wie es gelaufen ist.
Zitieren Sie dieses Tutorial
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 Aus dem Sequence Read Archive (SRA) des NCBI zu Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials). /schulungsmaterial/Themen/Variantenanalyse/tutorials/sars-cov-2/tutorial.html Online; Zugriff HEUTE
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}