El objetivo de este tutorial es presentarle el procesamiento de datos de secuenciación de próxima generación en Galaxy. Este tutorial utiliza una llamada de variante COVID-19 desde datos Illumina, pero no se trata de llamadas de variante per se.
Al completar este tutorial, sabrá:
- Cómo encontrar datos en SRA y transferir esta información a Galaxy
- Cómo realizar el procesamiento básico de datos NGS en Galaxy, incluido:
- Control de Calidad (QC) de Illumina de datos
- Mapeo
- la Eliminación de duplicados
- la Variante de la llamada
lofreq - la Variante de anotación
- el Uso de conjuntos de datos de colecciones
- la Importación de datos a Jupyter
### Agenda>> En este tutorial, vamos a cubrir:>> 1. TOC> {:toc}>{: .agenda} # # Dos rutas a través de este tutorecreamos dos trayectorias que puedes seguir a través de este tutorial.1. ** Trayectoria 1 * * – comience con el SRA de NCBI y busque las accesiones disponibles → Iniciar (#el archivo de lectura de secuencia) 2. ** Trayectoria 2**: evita el SRA de NCBI y comienza con Galaxy directamente. → Inicio (#back-in-galaxy) Recomendamos comenzar con * * Trayectoria 2**.# El archivo de lectura de secuencia (https://www.ncbi.nlm.nih.gov/sra) es el archivo principal de *lecturas sin ensamblar* para el (https://www.ncbi.nlm.nih.gov/). SRA es un excelente lugar para obtener los datos de secuenciación que subyacen a publicaciones y estudios.Este tutorial cubre cómo obtener datos de secuencia de SRA a Galaxy utilizando una conexión directa entre los dos.> # # # comment Comment>> También escuchará a SRA como el *Archivo de lectura corta*, su nombre original.> {:.comentario}## Se puede acceder a SRASRA directamente a través de su sitio web o a través del panel de herramientas en Galaxy.> # # # comment Comment>>Inicialmente, la opción del panel de herramientas para acceder a SRA solo existe en (https://usegalaxy.org/). El soporte para la conexión directa a SRA se incluirá en la versión 20.05 de Galaxy {:.comentario}> ### hands_on Manos: Explorar SRA Entrez>> 1. Ir a su Galaxy instancia de elección como uno de los (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) o cualquier otro. (Este tutorial utiliza usegalaxy.org).> 1. Si su historial no está vacío, inicie un nuevo historial (consulte (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) para obtener más información sobre los historiales de galaxias)> 1. ** Haga clic en * * ‘Obtener datos’ en la parte superior del panel de herramientas.> 1. ** Haga clic en * * ‘Servidor SRA’ en la lista de herramientas que se muestra en ‘Obtener datos’.> Esto le lleva el (https://www.ncbi.nlm.nih.gov/sra) also también puede comenzar directamente desde el SRA. Se muestra un cuadro de búsqueda en la parte superior de la página. Intenta buscar algo que te interese, como `delfín`, `riñón` o `riñón delfín` y luego **haz clic** en el botón `Buscar`.>> Esto devuelve una lista de *Experimentos SRA* que coinciden con su cadena de búsqueda. Los experimentos SRA, también conocidos como entradas *SRX*, contienen datos de secuencia de un experimento en particular, así como una explicación del experimento en sí y cualquier otro dato relacionado. Puede explorar los experimentos devueltos haciendo clic en su nombre. Consulte (https://www.ncbi.nlm.nih.gov/books / NBK56913/) en (https://www.ncbi.nlm.nih.gov/books/n/ helpsrakb/) para obtener más información.>> Cuando introduzca texto en el SRA cuadro de búsqueda, que está utilizando (https://www.ncbi.nlm.nih.gov/sra/docs/srasearch/). Entrez admite búsquedas de texto simples y búsquedas muy precisas que comprueban metadatos específicos y usan expresiones lógicas arbitrariamente complejas. Entrez le permite escalar sus búsquedas de básicas a avanzadas a medida que reduce sus búsquedas. La sintaxis de las búsquedas avanzadas puede parecer desalentadora, pero SRA proporciona un gráfico (https://www.ncbi.nlm.nih.gov/sra/advanced/) para generar la sintaxis específica. Y, como veremos a continuación, el Selector de ejecución de SRA proporciona una interfaz de usuario aún más amigable para reducir los datos seleccionados.>> Juegue con la interfaz SRA Entrez, incluido el generador de consultas avanzado, para ver si puede identificar un conjunto de experimentos SRA que sean relevantes para una de sus áreas de investigación.{: .hands_on}> # # # hands_on Hands-on: Genere una lista de experimentos coincidentes utilizando Entrez>> Ahora que tiene una familiaridad básica con SRA Entrez, encontremos las secuencias utilizadas en este tutorial.>> 1. Si aún no está allí, **navegue** de vuelta a (https://www.ncbi.nlm.nih.gov/sra> 1. ** Borra* * cualquier texto de búsqueda del cuadro de búsqueda.> 1. ** Escriba* * `sars-cov-2 `en el cuadro de búsqueda y **haga clic en**`Buscar’.> Esto devuelve una larga lista de experimentos SRA que coinciden con nuestra búsqueda, y esa lista es demasiado larga para usarla en un ejercicio tutorial. En este punto, podríamos usar el generador de consultas Entrez avanzado del que aprendimos anteriormente.> Pero no lo haremos. En su lugar, enviemos la lista de resultados *demasiado larga para un tutorial* que tenemos al Selector de ejecución de SRA, y use su interfaz más amigable para limitar nuestros resultados.>> !(../../ images / sra_entrez.png){: .hands_on}> # # # hands_on Hands-on: Vaya de Entrez al Selector de ejecución SRA>> Vea los resultados como una tabla interactiva ampliada con el selector de ejecución.>> 1. Haga clic en Enviar resultados para Ejecutar el selector, que aparece en un cuadro en la parte superior de los resultados de búsqueda.>> !(../../ images / sra_entrez_result.png)>>> ### tip Lo que si no vi la carrera Selector de Enlace?>>>> Usted puede haber notado este texto anterior cuando se fueron a explorar Motor de búsqueda. Este texto solo aparece una parte del tiempo, cuando el número de resultados de búsqueda cae dentro de una ventana bastante amplia. No lo verá si solo tiene unos pocos resultados, y no lo verá si tiene más resultados de los que el Selector de ejecución puede aceptar.>>>> *Usted necesita para llegar a Correr Selector de enviar los resultados a la Galaxia.* ¿ Qué pasa si no tiene suficientes resultados para activar que se muestre este enlace? En ese caso, llame al Selector de ejecución * * haciendo clic en * * en el menú desplegable «Enviar a» en la parte superior derecha del panel de resultados. Para llegar al Selector de ejecución, * * seleccione * * `Selector de ejecución ‘y luego * * haga clic en * * el botón ‘Ir’.>!(../../ images / sra_entrez_send_to.png)> {: .tip}>>> 1. ** Haga clic en * * ‘Enviar resultados al selector de ejecución’ en la parte superior del panel de resultados de búsqueda. (Si no ves este enlace, consulta el comentario directamente arriba.){: .hands_on}## SRA Run Selector Aprendimos anteriormente cómo reducir los resultados de búsqueda utilizando la sintaxis avanzada de Entrez. Sin embargo, no aprovechamos ese poder cuando estábamos en Entrez. En su lugar, usamos una búsqueda simple y luego enviamos todos los resultados al Selector de ejecución. Todavía no tenemos la lista (corta) de resultados sobre los que queremos ejecutar el análisis. *¿Qué estamos haciendo?* Estamos usando Entrez y el Selector de ejecución cómo están diseñados para ser utilizados: * Utilice la interfaz Entrez para reducir sus resultados a un tamaño que el Selector de ejecución pueda consumir. * Envíe esos resultados de Entrez al Selector de ejecución de SRA * Utilice la interfaz mucho más amigable del Selector de ejecución a 1. Comprender más fácilmente los datos que tenemos 1. Reduce esos resultados usando ese conocimiento.> ### comment Ejecutar Selector es a la vez más y menos de Entrez>> Ejecutar Selector que puede hacer la mayoría, pero no todos de qué Motor de búsqueda de sintaxis puede hacer. El selector de ejecución utiliza la tecnología de búsqueda facetada que es fácil de usar y potente, pero que tiene límites inherentes. Específicamente, Entrez funcionará mejor al buscar atributos que tengan decenas, cientos o miles de valores diferentes. El selector de ejecución funcionará mejor buscando atributos con menos de 20 valores diferentes. Afortunadamente, eso describe la mayoría de las búsquedas.{: .comentario}La ventana del Selector de ejecución se divide en varios paneles:* **`Lista de filtros`**: En la esquina superior izquierda. Aquí es donde refinaremos nuestra búsqueda.* * * ` Seleccionar’**: Un resumen de lo que se pasó inicialmente al Selector de ejecución, y cuánto de eso hemos seleccionado hasta ahora. (Y hasta ahora, no hemos seleccionado ninguno de ellos.) También tenga en cuenta el tentador, pero todavía gris, botón `Galaxia`.** * `Elementos encontrados x` * * Inicialmente, esta es la lista de elementos enviados al Selector de Ejecución desde Entrez. Esta lista se reducirá a medida que le apliquemos filtros.!(../../ images / sra_run_selector.png)> # # # comentario ¿Por qué aumentó el número de elementos encontrados*?* >> Recuerde que la interfaz Entrez enumera los experimentos SRA (entradas SRX). Ejecutar listas selectoras * ejecuciones * – secuenciación de conjuntos de datos – y hay* una o más * ejecuciones por experimento. Tenemos los mismos datos que antes, ahora los estamos viendo con más detalle.{: .comentario}La `Lista de filtros ‘ en la parte superior izquierda muestra columnas en nuestros resultados que tienen valores numéricos continuos, o 10 o menos (puede cambiar este número) valores distintos en ellos. ** Desplácese* * hacia abajo por la lista, seleccione algunos de los filtros. Cuando se selecciona un filtro, aparece un cuadro *valores* a continuación, con una lista de opciones para este filtro y el número de ejecuciones con cada opción. Estos valores / opciones se extraen de los metadatos del conjunto de datos. Intente * * seleccionar * * algunos filtros de sonido interesantes y luego * * seleccionar * * una o más opciones para cada filtro. Intente * * deseleccionar * * opciones y filtros. Al hacer esto, el número de resultados encontrados disminuirá o aumentará.> # # # tip Tip: Use filtros para comprender mejor los datos>> Los filtros son la forma de reducir los conjuntos de datos que se consideran para enviar a Galaxy, pero también son una excelente manera de comprender sus datos:> En primer lugar, seleccionar un filtro es una forma fácil de ver el rango de valores en una columna. Es posible que no pueda (https://www.google.com/search?q=sra+sirs_outcome), pero es posible que pueda averiguarlo viendo qué valores contiene.> En segundo lugar, puede explorar cómo se relacionan las diferentes columnas entre sí. ¿Existe una relación entre los valores «sirs_outcome» y los valores «disease_stage»?{: .tip}> ### hands_on Manos: Estrecho de los resultados mediante Ejecutar Selector>> 1. Si tiene algún filtro activado, **deseleccione** los filtros.> Una vez que haya hecho esto, no aparecerá ningún cuadro de *valores* debajo de la `Lista de filtros`.> 2. ** Copie y pegue * * esta cadena de búsqueda en el cuadro de búsqueda ‘Elementos encontrados’.>> SRR11772204 O SRR11597145 O SRR11667145>> Este recogidos a mano conjunto de pistas de los límites de nuestros resultados a 3 pistas de diferente distribución geográfica.{: .hands_on} Esto reduce la lista de ‘Objetos encontrados’ de decenas de miles de corridas a 3 corridas (¡un número manejable para un tutorial!). Pero aún no hemos terminado con el Selector de ejecución. Ten en cuenta que el botón «Galaxia» sigue en gris. Hemos reducido nuestras opciones, pero aún no hemos seleccionado nada para enviar a Galaxy.Es posible seleccionar cada ejecución restante haciendo clic en** en la marca de verificación en la parte superior de la primera columna. Puede deseleccionar todo * * haciendo clic* * en la ‘X’.> ### hands_on Manos: Seleccionar pistas y enviar a la Galaxia>> 1. Seleccione todas las carreras * * haciendo clic en* * en la ‘X’.> Y ahora, el botón ‘Galaxy’ está activo.> 1. ** Haga clic en * * el botón` Galaxy ‘en la sección` Seleccionar’ en la parte superior de la página.{: .hands_on} # # De vuelta en Galaxycuando hacemos clic en ‘Galaxy’ en el Selector de ejecución suceden varias cosas. Primero, lanza una nueva pestaña o ventana del navegador que se abre en Galaxy. Verá el * gran recuadro verde * que indica que el apretón de manos entre SRA y Galaxy fue exitoso y luego verá un nuevo trabajo `SRA` en su panel de historial. Esta casilla puede comenzar como gris / pendiente, lo que indica que la transferencia aún no ha comenzado, o puede ir directamente a amarillo / en ejecución o a verde / listo.> # # # hands_on Hands-on: Examine el nuevo conjunto de datos SRA>> 1. Una vez completada la transferencia` SRA’, **haga clic en** en el icono galaxy-eye (ojo) del conjunto de datos.>> Esto muestra el conjunto de datos en la Galaxia del panel central.{: .hands_on}El conjunto de datos `SRA ‘ no son datos de secuencia, sino más bien *metadatos* que usaremos para obtener datos de secuencia de SRA. Estos metadatos reflejan la información que vimos en la sección `Elementos encontrados` del Selector de ejecución. Los metadatos no son los datos finales que estamos buscando de SRA, pero tener todos esos metadatos a menudo es útil en los pasos de análisis posteriores.Ahora usemos esos metadatos para obtener los datos de secuencia de SRA. SRA proporciona herramientas para extraer todo tipo de información, incluidos los datos de secuencia en sí. La herramienta Galaxy ‘Lectura de descarga y extracción más rápida en FASTQ’ se basa en la utilidad SRA (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump), y hace precisamente eso.– >
- Encuentra los datos necesarios en SRA
- hands_on Hands-on: Descripción de la tarea
- comentario Comentario
- Procesar y filtrar SraRunInfo.archivo csv en Galaxy
- hands_on Hands-on: Subir SraRunInfo.archivo csv en Galaxy
- comentario Tenga cuidado con los cortes
- hands_on Hands-on: Creación de un subconjunto de datos
- consejo: Encontrar herramientas
- Descargar datos de secuenciación con Lecturas de Descarga y Extracción más rápidas en FASTQ
- hands_on Hands-on: Descripción de la tarea
- ¿Ahora qué?
- Análisis de variaciones de los datos de secuenciación del SARS-Cov-2
- comenta el usegalaxy.* Proyecto de análisis de COVID-19
- Obtener los datos del genoma de referencia
- hands_on Hands-on: Obtenga los datos del genoma de referencia
- Sugerencia: Importar a través de enlaces
- Recortar adaptadores con fastp
- hands_on Hands-on: Descripción de la tarea
- Alineación con mapa con BWA-MEM
- hands_on Hands-on: Alinee las lecturas de secuenciación con el mapa de referencia del genoma
- Eliminar duplicados con MarkDuplicates
- hands_on Hands-on: Eliminar duplicados de PCR
- Generar estadísticas de alineación con Samtools stats
- hands_on Hands-on: Generar estadísticas de alineación
- Realinear lecturas con lofreq viterbi
- hands_on Hands-on: Realinear lecturas alrededor de indels
- Agregar indel cualidades con lofreq Insertar indel cualidades
- hands_on Hands-on: Agregar indel cualidades
- Llamada utilizando las Variantes lofreq Llamada variantes
- hands_on Hands-on: Variantes de llamada
- Anotar efectos de variantes con SnpEff eff:
- hands_on Hands-on: Anotar efectos de variantes
- Crear tabla de variantes usando campos de extracción SnpSift
- hands_on Hands-on: Crear tabla de variantes
- Resumir datos con MultiQC
- hands_on Hands-on: Resumir datos
- Conclusión
- keypoints puntos Clave
- Preguntas Frecuentes
- Literatura útil
- Feedback
- Citando este tutorial
- details BibTeX
Encuentra los datos necesarios en SRA
Primero necesitamos encontrar un buen conjunto de datos con el que jugar. La Secuencia de Lectura de Archivo (SRA) es el principal archivo de desmontado lee operados por los Institutos Nacionales de Salud (NIH). SRA es un excelente lugar para obtener los datos de secuenciación que subyacen a publicaciones y estudios. Vamos a hacer eso:
hands_on Hands-on: Descripción de la tarea
- Vaya a la página SRA de NCBI apuntando su navegador a https://www.ncbi.nlm.nih.gov/sra
- En el cuadro de búsqueda ingrese
SARS-CoV-2 Patient Sequencing From Partners / MGH(Alternativamente, simplemente haga clic en este enlace)
- La página web mostrará un gran número de conjuntos de datos SRA (en el momento de escribir esto había 2,223). Estos son datos de un estudio que describe el análisis del SARS-CoV-2 en el área de Boston.
- Descargue metadatos que describan estos conjuntos de datos haciendo clic en Enviar a: desplegable
- Seleccionando
File- Cambiando el formato a
RunInfo- Haciendo clic en Crear archivo Aquí está cómo debería verse:
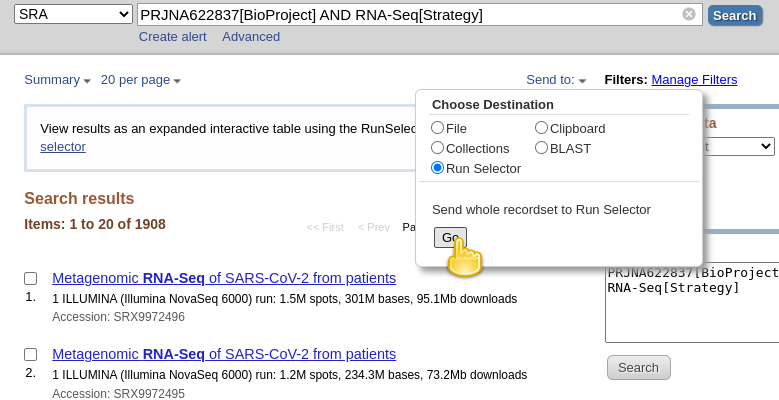
- Esto crearía un gran
SraRunInfo.csvarchivoDownloadscarpeta.
Ahora que hemos descargado este archivo, podemos ir a una instancia de Galaxy y comenzar a procesarlo.
comentario Comentario
Tenga en cuenta que el archivo que acabamos de descargar no está secuenciando los datos en sí. Más bien, son metadatos que describen las propiedades de las lecturas de secuenciación. Filtraremos esta lista a solo unas pocas accesiones que se usarán en el resto de este tutorial.
Procesar y filtrar SraRunInfo.archivo csv en Galaxy
hands_on Hands-on: Subir SraRunInfo.archivo csv en Galaxy
- Vaya a la instancia de Galaxy de su elección, como una de las usegalaxy.org, usegalaxy.eu, usegalaxy.org.au o cualquier otra. (Este tutorial utiliza usegalaxy.org).
- Haga clic en el botón Cargar datos:
- En el cuadro de diálogo que aparecerá, haga clic en el botón «Elegir archivos locales»:

- Buscar y seleccionar
SraRunInfo.csvarchivo de su computadora- Haga clic en el botón Inicio
- Cerrar el diálogo pulsando Botón Cerrar
- Ahora puede ver el contenido de este archivo haciendo clic en el icono galaxy-eye (ojo). Verá que este archivo contiene mucha información sobre las accesiones SRA individuales. En este estudio, cada acceso corresponde a un paciente individual cuyas muestras fueron secuenciadas.
Galaxy puede procesar más de 2.000 conjuntos de datos, pero para que este tutorial sea soportable, necesitamos seleccionar un subconjunto más pequeño. En particular, nuestra experiencia previa con estos datos muestra dos conjuntos de datos interesantes SRR11954102 y SRR12733957. Así que, vamos a sacarlos.
comentario Tenga cuidado con los cortes
La sección Práctica a continuación utiliza la herramienta Cortar. Hay dos herramientas de corte en Galaxy debido a razones históricas. Este ejemplo utiliza la herramienta con el nombre completo Cortar columnas de una tabla (cortar). Sin embargo, la misma lógica se aplica a la otra herramienta. Simplemente tiene una interfaz ligeramente diferente.
hands_on Hands-on: Creación de un subconjunto de datos
- herramienta Buscar «Seleccionar líneas que coincidan con una expresión» en la sección Filtrar y Ordenar del panel de herramientas.
consejo: Encontrar herramientas
Galaxy puede tener una cantidad abrumadora de herramientas instaladas. Para encontrar una herramienta específica, escriba el nombre de la herramienta en el cuadro de búsqueda del panel de herramientas para encontrar la herramienta.
- Asegúrese de que el conjunto de datos
SraRunInfo.csvque acabamos de cargar esté listado en el campo archivo param» Seleccionar líneas de » del formulario de herramienta.- En el campo» el patrón «introduzca la siguiente expresión →
SRR12733957|SRR11954102. Estos son dos accesos que queremos encontrar separados por el símbolo de tubería|. El|significaor: encontrar líneas que contienenSRR12733957oSRR11954102.- Haga clic en el botón
Execute.- Esto generará un archivo que contiene dos líneas (bueno well una línea también se usa como encabezado, por lo que aparecerá que el archivo tiene tres líneas. Está bien.)
- Corte la primera columna del archivo utilizando la herramienta» Cortar», que encontrará en la sección Manipulación de texto del panel de herramientas.
- Asegúrese de que el conjunto de datos producido por el paso anterior esté seleccionado en el campo» Archivo a cortar » del formulario de herramienta.
- Cambio «Delimitado por»
Comma- En la «Lista de campos» seleccione
Column: 1.- Hit
ExecuteEsto producirá un archivo de texto con solo dos líneas:SRR12733957SRR11954102
Ahora que tenemos identificadores de conjuntos de datos que queremos necesidad de descargar los datos de secuenciación reales.
Descargar datos de secuenciación con Lecturas de Descarga y Extracción más rápidas en FASTQ
hands_on Hands-on: Descripción de la tarea
- Lecturas de descarga y extracción más rápidas en la herramienta FASTQ con los siguientes parámetros:
- «seleccionar tipo de entrada»:
List of SRA accession, one per line
- El archivo de parámetros «lista de adhesión sra» debe apuntar a la salida de la herramienta «Cortar» del paso anterior.
- Haga clic en el botón
Execute. Esto ejecutará la herramienta, que recupera los conjuntos de datos de lectura de secuencia para las ejecuciones que se enumeraron en el conjunto de datosSRA. Puede llevar algún tiempo. Así que este puede ser un buen momento para tomar café.- Se crean varias entradas en el panel historial al enviar este trabajo:
Pair-end data (fasterq-dump): Contiene conjuntos de datos de extremo emparejado (si está disponible)Single-end data (fasterq-dump)Contiene conjuntos de datos de extremo único (si está disponible)Other data (fasterq-dump)Contiene conjuntos de datos no emparejados (si está disponible)fasterq-dump logContiene Información sobre la ejecución de la herramienta
Los tres primeros elementos son en realidad colecciones de conjuntos de datos. Las colecciones en Galaxy son agrupaciones lógicas de conjuntos de datos que reflejan las relaciones semánticas entre ellos en el experimento / análisis. En este caso, la herramienta crea una colección separada para lecturas de extremos emparejados, lecturas individuales y otras.Consulte los tutoriales de colecciones para obtener más información.
Explore las colecciones haciendo clic primero en el nombre de la colección en el panel historial. Esto le lleva dentro de la colección y le muestra los conjuntos de datos que contiene. A continuación, puede volver al nivel externo de su historial.
Una vez que fasterq termine de transferir datos (todos los cuadros están verdes / listos), estamos listos para analizarlos.
¿Ahora qué?
Ahora puede analizar los datos recuperados utilizando cualquier herramienta de análisis de secuencias y flujos de trabajo en Galaxy. SRA contiene datos de respaldo para cada tipo imaginable de experimento *-seq.
Si ejecutó este tutorial, pero recuperó conjuntos de datos que le interesaban, consulte el resto de la biblioteca GTN para obtener ideas sobre cómo analizar en Galaxy.
Sin embargo, si recuperó los conjuntos de datos utilizados en los ejemplos anteriores de este tutorial, estará listo para ejecutar el análisis de variantes del SARS-CoV-2 a continuación.
Análisis de variaciones de los datos de secuenciación del SARS-Cov-2
En esta parte del tutorial realizaremos llamadas de variantes y análisis básico de los conjuntos de datos descargados anteriormente. Comenzaremos descargando la secuencia de referencia de Wuhan-Hu-1 SARS-CoV-2, luego ejecutaremos el recorte del adaptador, la alineación y la llamada de variantes y, finalmente, veremos la distribución geográfica de algunas de las variantes encontradas.
comenta el usegalaxy.* Proyecto de análisis de COVID-19
Este tutorial utiliza un subconjunto de los datos y recorre la sección de Análisis de variables de la covid19.proyecto galaxia.org.Los datos para covid19.galaxyproject.org se actualiza continuamente a medida que se publican nuevos conjuntos de datos.
Obtener los datos del genoma de referencia
Los datos del genoma de referencia para hoy son para el SARS-CoV-2, «Síndrome respiratorio agudo severo coronavirus 2 aislado Wuhan-Hu-1, genoma completo», con el ID de acceso NC_045512.2.
Estos datos están disponibles en Zenodo utilizando el siguiente enlace.
hands_on Hands-on: Obtenga los datos del genoma de referencia
Importe el siguiente archivo en su historial:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzSugerencia: Importar a través de enlaces
- Copie la ubicación del enlace
- Abra el Administrador de carga de Galaxy (carga de Galaxy en la parte superior derecha del panel de herramientas)
- Seleccione Pegar/Buscar Datos
- Pegue el enlace en el campo de texto
- Presione Inicio
- Cierre la ventana Por defecto, Galaxy usa la URL como nombre, por lo que cambie el nombre de los archivos con un nombre más útil.
Recortar adaptadores con fastp
Eliminar adaptadores de secuenciación mejora las alineaciones y las llamadas de variantes. la herramienta fastp puede detectar automáticamente adaptadores de secuenciación ampliamente utilizados.
hands_on Hands-on: Descripción de la tarea
- Herramienta fastp con los siguientes parámetros:
- «Lecturas de un solo extremo o emparejadas»:
Paired Collection
- archivo param»Seleccionar colección(s) emparejada (s)»:
list_paired(salida de lecturas de descarga y extracción más rápidas en la herramienta FASTQ)- En» Opciones de salida»:
- » Informe de salida JSON»:
Yes
Alineación con mapa con BWA-MEM
La herramienta BWA-MEM es un alineador de secuencias ampliamente utilizado para conjuntos de datos de secuenciación de lectura corta, como los que analizamos en este tutorial.
hands_on Hands-on: Alinee las lecturas de secuenciación con el mapa de referencia del genoma
- con la herramienta BWA-MEM con los siguientes parámetros:
- » ¿Seleccionará un genoma de referencia de su historial o utilizará un índice incorporado?»:
Use a genome from history and build index
- archivo param «Utilice el siguiente conjunto de datos como secuencia de referencia»:
output(Conjunto de datos de entrada)- «Lecturas de extremo único o emparejado»:
Paired Collection
- archivo param «Seleccionar una colección emparejada»:
output_paired_coll(salida de la herramienta fastp)- «¿Establecer información de grupos de lectura?»:
Do not set- «Seleccionar modo de análisis»:
1.Simple Illumina mode
Eliminar duplicados con MarkDuplicates
La herramienta MarkDuplicates elimina secuencias duplicadas que se originan en la preparación de la biblioteca artefactos y artefactos de secuenciación. Es importante eliminar estas secuencias de artefactos para evitar la sobrerepresentación artificial de una sola molécula.
hands_on Hands-on: Eliminar duplicados de PCR
- Herramienta de marcado de duplicados con los siguientes parámetros:
- archivo param «Seleccionar conjunto de datos SAM/BAM o colección de conjuntos de datos»:
bam_output(salida del mapa con la herramienta BWA-MEM)- «Si es verdadero, no escriba duplicados en el archivo de salida en lugar de escribirlos con los indicadores apropiados»:
Yes
Generar estadísticas de alineación con Samtools stats
Después del paso de marcado duplicado anterior, podemos generar estadísticas sobre la alineación que hemos generado.
hands_on Hands-on: Generar estadísticas de alineación
- Herramienta de estadísticas Samtools con los siguientes parámetros:
- archivo param «archivo BAM»:
outFile(herramienta de salida de duplicados de marcas)- «Establecer distribución de cobertura»:
No- «Salida»:
One single summary file- «Filtrar por banderas SAM»:
Do not filter- «Usar una secuencia de referencia»:
No- «Filtrar por regiones»:
No
Realinear lecturas con lofreq viterbi
La herramienta Realinear lecturas corrige los desalineamientos alrededor de inserciones y eliminaciones. Esto es necesario para detectar con precisión las variantes.
hands_on Hands-on: Realinear lecturas alrededor de indels
- Realinear lecturas con la herramienta lofreq con los siguientes parámetros:
- archivo de parámetros «Lecturas para realinear»:
outFile(salida de la herramienta MarkDuplicates)- «Elija la fuente para el genoma de referencia»:
History
- archivo param» Referencia»:
output(Conjunto de datos de entrada)- En» Opciones avanzadas»:
- » ¿Cómo manejar las cualidades base de 2?»:
Keep unchanged
Agregar indel cualidades con lofreq Insertar indel cualidades
Este paso agrega indel cualidades en nuestra alineación de archivo. Esto es necesario para llamar a variantes utilizando variantes de llamada con la herramienta lofreq
hands_on Hands-on: Agregar indel cualidades
- Insertar indel cualidades con lofreq herramienta con los siguientes parámetros:
- param-archivo «Lee»:
realigned(salida de Realinear lee herramienta)- «Indel método de cálculo»:
Dindel
- «Seleccione la fuente para el genoma de referencia»:
History
- param-archivo de «Referencia»:
output(conjunto de datos de Entrada)
Llamada utilizando las Variantes lofreq Llamada variantes
ahora Estamos listos para llamar variantes.
hands_on Hands-on: Variantes de llamada
- Variantes de llamada con la herramienta lofreq con los siguientes parámetros:
- Archivo param «Lecturas de entrada en formato BAM»:
output(salida de la herramienta Insert indel qualities)- «el genoma de referencia»:
History
- archivo param «Referencia»:
output(Conjunto de datos de entrada)- «Variantes de llamada a través»:
Whole reference- «Tipos de variantes a llamar»:
SNVs and indels- «Variantes de los parámetros de llamada»:
Configure settings
- En la «Cobertura»:
- «Mínima cobertura»:
50- En «la Base de llamada»:
- «Mínimo baseq»:
30- «Mínimo baseq para las bases»:
30- en «asignación de qualityy
20- «variante de los parámetros de los filtros»:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
El resultado de este paso es una colección de archivos VCF que puede ser visualizado en un navegador del genoma.
Anotar efectos de variantes con SnpEff eff:
Ahora anotaremos las variantes que llamamos en el paso anterior con el efecto que tienen en el genoma del SARS-CoV-2.
hands_on Hands-on: Anotar efectos de variantes
- SnpEff eff: herramienta con los siguientes parámetros:
- archivo de parámetros «Cambios de secuencia (SNPs, MNPs, InDels)»:
variants(herramienta de variantes de salida de llamadas)- «Formato de salida»:
VCF (only if input is VCF)- «Crear informe CSV, útil para análisis descendentes (-csvStats)»:
Yes- «Opciones de anotación»: `
- «Filtrar salida»: `
- «Filtrar efectos específicos»:
No
La salida de este paso es un archivo VCF con efectos variantes añadidos.
Crear tabla de variantes usando campos de extracción SnpSift
Ahora seleccionaremos varios efectos del VCF y crearemos un archivo tabular que sea más fácil de entender para los humanos.
hands_on Hands-on: Crear tabla de variantes
- Herramienta de extracción de campos SnpSift con los siguientes parámetros:
- archivo param «Archivo de entrada de variante en formato VCF»:
snpeff_output(salida de SnpEff eff: herramienta)- «Campos a extraer»:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- «separador de campos múltiples»:
,- «texto de campo vacío»:
.
Podemos inspeccionar los archivos de salida y ver si las variantes de este archivo también se describen en un cuaderno observable que muestra distribución de secuencias de variantes del SARS-CoV-2
Las variantes interesantes incluyen la variante C a T en la posición 14408 (14408C/T) en SRR11772204, 28144T/C en SRR11597145 y 25563G/T en SRR11667145.
Resumir datos con MultiQC
Ahora resumiremos nuestro análisis con MultiQC, que genera un hermoso informe para nuestros datos.
hands_on Hands-on: Resumir datos
- Herramienta MultiQC con los siguientes parámetros:
- En «Resultados»:
- param-repetir» Insertar resultados «
- » ¿Qué herramienta se utilizó para generar registros?»:
fastp
- archivo de parámetros «Salida de fastp»:
report_json(salida de la herramienta fastp)- param-repetir «Insertar resultados»
- «¿Qué herramienta se utilizó para generar registros?»:
Samtools
- En «Salida de Samtools»:
- repetición de param» Insertar salida de Samtools «
- » ¿Tipo de salida de Samtools?»:
stats
- archivo de parámetros «Salida de estadísticas de Samtools»:
output(salida de Samtools herramienta de estadísticas)- param-repita «Insertar» Resultados de la
- «la herramienta Que se utilizó generar registros?»:
Picard
- En «Salida Picard»:
- repetición de param» Insertar salida Picard «
- » Tipo de salida Picard?»:
Markdups- archivo de parámetros «salida Picard»:
metrics_file(salida de MarkDuplicates herramienta)- param-repita «Insertar» Resultados de la
- «la herramienta Que se utilizó generar registros?»:
SnpEff
- param-archivo de Salida «de SnpEff»:
csvFile(salida de SnpEff fep: herramienta)
Conclusión
Felicitaciones, ahora sabes cómo importar datos de la secuencia de la SRA y de cómo ejecutar un ejemplo de análisis de estos conjuntos de datos.
keypoints puntos Clave
la Secuencia de datos en el SRA se pueden importar directamente en Galaxy
Preguntas Frecuentes
Tiene preguntas acerca de este tutorial? Consulta la página de preguntas frecuentes del tema de Análisis de variantes para ver si tu pregunta aparece en la lista. Si no es así, haga su pregunta en el Canal GTN Gitter o en el Foro de Ayuda Galaxy
Literatura útil
Puede encontrar más información, incluidos enlaces a documentación y publicaciones originales, sobre las herramientas, las técnicas de análisis y la interpretación de los resultados descritos en este tutorial aquí.
Feedback
¿Utilizó este material como instructor? Siéntase libre de darnos su opinión sobre cómo fue.
Citando este tutorial
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 Del Archivo de Lectura de secuencias de NCBI (SRA) al análisis de variantes de Galaxy: SARS-CoV-2 (Materiales de capacitación de Galaxy). / material de capacitación/temas/análisis de variantes/tutoriales/sars-cov-2 / tutorial.html en línea; se accede HOY
- Batut et al., 2018 Entrenamiento de Análisis de Datos Impulsado por la Comunidad para Sistemas Celulares de Biología 10.1016 / j. cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}