Forma de dosificación: inyección
Revisado médicamente por Drugs.com. Última actualización el 22 de diciembre de 2020.
- Efectos Secundarios
- Dosis
- Profesional
- Interacciones
- el Embarazo
- Más
SÓLO Rx.
Solución intravenosa estéril
- Inamrinona Descripción
- Inamrinona-Farmacología clínica
- Farmacocinética
- Farmacodinámica
- Indicaciones y uso de Inamrinona
- Contraindicaciones
- Advertencias
- Precauciones
- General
- Pruebas de laboratorio
- Interacciones medicamentosas
- Interacciones químicas
- Carcinogénesis, Mutagénesis, Alteración de la fertilidad
- Embarazo
- Madres lactantes
- Uso pediátrico
- Reacciones Adversas
- Manejo de las Reacciones Adversas
- Sobredosis
- Dosis y administración de Inamrinona
- Interacciones químicas
- Cómo se suministra la Inamrinona
- VIAL LABEL
- More about inamrinone
- Related treatment guides
Inamrinona Descripción
La inyección de inamrinona USP representa una nueva clase de agentes inotrópicos cardíacos distintos de los glucósidos digitálicos o las catecolaminas. El lactato de inamrinona se designa químicamente como 5-Amino-6(1H)-un 2-hidroxipropanato y tiene la siguiente estructura:
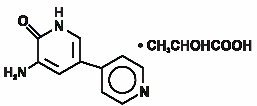
La inamrinona es un compuesto cristalino de color amarillo pálido con un peso molecular de 187,20 y una fórmula molecular de C10H9N3O. Cada mol de ácido láctico tiene un peso molecular de 90,08 y una fórmula empírica de C3H6O3. Las solubilidades de Inamrinona a pH de 4,1, 6,0 y 8,0 son de 25, 0,9 y 0,7 mg/ml, respectivamente.
La inyección de inamrinona es una solución estéril de color amarillo transparente disponible en viales de 20 mL para administración intravenosa. Cada ml contiene lactato de inamrinona equivalente a 5 mg de Inamrinona y 0,25 mg de metabisulfito de sodio añadido como conservante en Agua para preparaciones inyectables. Todas las dosis expresadas en el prospecto se expresan en términos de la base, inamrinona. El pH se ajusta entre 3,2 y 4,0 con ácido láctico o hidróxido de sodio. La concentración total de ácido láctico puede variar entre 5 mg y 7,5 mg.
Inamrinona-Farmacología clínica
La inamrinona es un agente inotrópico positivo con actividad vasodilatadora, diferente en estructura y modo de acción de los glucósidos digitálicos o de las catecolaminas.
El mecanismo de sus efectos inotrópicos y vasodilatadores no ha sido completamente dilucidado.
Con respecto a su efecto inotrópico, la evidencia experimental indica que no es un agonista beta-adrenérgico. Inhibe la actividad de la fosfodiesterasa del adenosina monofosfato cíclico miocárdico (c-AMP) y aumenta los niveles celulares de c-AMP. A diferencia de la digitálica, no inhibe la actividad de la adenosina trifosfatasa de sodio y potasio.
Con respecto a su actividad vasodilatadora, la inamrinona reduce la poscarga y la precarga por su efecto relajante directo sobre el músculo liso vascular.
Farmacocinética
Tras la inyección intravenosa en bolo (1 a 2 minutos) de 0,68 mg/kg a 1,2 mg/kg a voluntarios normales, la inamrinona tuvo un volumen de distribución de 1,2 litros / kg, y tras una semivida en fase distributiva de aproximadamente 4,6 minutos en plasma, tuvo una semivida de eliminación terminal aparente de primer orden de aproximadamente 3,6 horas. En pacientes con insuficiencia cardíaca congestiva que recibieron perfusiones de inamrinona, la semivida de eliminación terminal aparente de primer orden fue de aproximadamente 5,8 horas.
En un estudio se ha demostrado que la inamrinona se une del 10% al 22% a las proteínas plasmáticas humanas mediante ultrafiltración in vitro, y en otro estudio se une del 35% al 49% mediante ultrafiltración o diálisis de equilibrio.
La principal vía de excreción en el hombre es a través de la orina en forma de inamrinona y varios metabolitos (N-glicolilo, N-acetato, O-glucurónido y N-glucurónido). En voluntarios sanos, aproximadamente el 63% de una dosis oral de Inamrinona marcada con 14C se excretó en la orina durante un periodo de 96 horas. En las primeras 8 horas, el 51% de la radiactividad en la orina fue inamrinona con un 5% como N-acetato, un 8% como N-glicolato y menos del 5% para cada glucurónido. Aproximadamente el 18% de la dosis administrada se excretó en las heces en 72 horas.
En un estudio intravenoso no radiactivo de 24 horas, del 10% al 40% de la dosis se excretó en orina como Inamrinona inalterada con el metabolito N-acetil representando menos del 2% de la dosis.
En pacientes con insuficiencia cardíaca congestiva, después de una dosis de carga en bolo, los niveles plasmáticos en estado estacionario de aproximadamente 2,4 mcg/ml se pudieron mantener mediante una infusión de 5 mcg/kg/min a 10 mcg/kg/min. En algunos pacientes con insuficiencia cardíaca congestiva, con perfusión renal y hepática comprometida asociada, es posible que los niveles plasmáticos de Inamrinona aumenten durante el período de infusión; por lo tanto, en estos pacientes, puede ser necesario monitorear la respuesta hemodinámica y/o el nivel del fármaco. Las principales medidas de respuesta del paciente incluyen el índice cardíaco, la presión de cuña capilar pulmonar, la presión venosa central y su relación con las concentraciones plasmáticas. Además, las mediciones de la presión arterial, la producción de orina y el peso corporal pueden resultar útiles, al igual que síntomas clínicos como la ortopnea, la disnea y la fatiga.
Farmacodinámica
En pacientes con función miocárdica deprimida, la inamrinona produce un aumento rápido del gasto cardíaco debido a sus acciones inotrópicas y vasodilatadoras.
Después de una dosis única intravenosa en bolo de Inamrinona de 0.75 mg / kg a 3 mg/kg en pacientes con insuficiencia cardíaca congestiva, se producen aumentos máximos del gasto cardíaco relacionados con la dosis (de aproximadamente el 28% a 0,75 mg/kg a aproximadamente el 61% a 3 mg/kg). El efecto máximo se produce en 10 minutos con todas las dosis. La duración del efecto depende de la dosis, con una duración de aproximadamente 1/2 hora a 0,75 mg / kg y aproximadamente 2 horas a 3 mg/kg.
En el mismo rango de dosis, la presión de cuña capilar pulmonar y la resistencia periférica total muestran disminuciones relacionadas con la dosis (disminuciones máximas medias del 29% en la presión de cuña capilar pulmonar y del 29% en la resistencia vascular sistémica). A dosis de hasta 3 mg / kg se han observado disminuciones de la presión diastólica (hasta un 13%) relacionadas con la dosis. La presión arterial media disminuye (9,7%) a una dosis de 3 mg/kg. La frecuencia cardíaca generalmente no cambia.
Los cambios en los parámetros hemodinámicos se mantienen durante la infusión intravenosa continua y durante varias horas después.
La inamrinona es eficaz en pacientes completamente digitalizados sin causar signos de toxicidad cardíaca por glucósidos. Sus efectos inotrópicos son aditivos a los de la digitálica. En los casos de aleteo/fibrilación auricular, es posible que la inamrinona aumente la tasa de respuesta ventricular debido a su leve realce de la conducción A/V. En estos casos, se recomienda el tratamiento previo con digitálicos.
Se ha observado mejoría de la función ventricular izquierda y alivio de la insuficiencia cardíaca congestiva en pacientes con cardiopatía isquémica. La mejoría se ha producido sin inducir síntomas o signos electrocardiográficos de isquemia miocárdica.
Con frecuencia cardíaca y presión arterial constantes, los aumentos en el gasto cardíaco ocurren sin aumentos medibles en el consumo de oxígeno miocárdico o cambios en la diferencia de oxígeno arteriovenoso.
La actividad inotrópica se mantiene después de dosis intravenosas repetidas de Inamrinona. La administración de inamrinona produce beneficios hemodinámicos y sintomáticos en pacientes que no se controlan satisfactoriamente con la terapia convencional con diuréticos y glucósidos cardíacos.
Indicaciones y uso de Inamrinona
La inyección de inamrinona es para el manejo a corto plazo de la insuficiencia cardíaca congestiva. Debido a la experiencia limitada y al potencial de efectos adversos graves (ver REACCIONES ADVERSAS), la inamrinona debe utilizarse únicamente en pacientes que puedan ser estrechamente monitorizados y que no hayan respondido adecuadamente a digitálicos, diuréticos y/o vasodilatadores. La experiencia con Inamrinona intravenosa en ensayos controlados no supera las 48 horas de bolos repetidos y/o perfusiones continuas.
Tanto si se administra por vía oral, por vía intravenosa continua o por vía intravenosa intermitente, en ensayos controlados no se ha demostrado que la inamrinona ni ningún otro inotrópico dependiente de AMP cíclico sean seguros o eficaces en el tratamiento a largo plazo de la insuficiencia cardíaca congestiva. En ensayos controlados de terapia oral crónica con varios de estos fármacos (incluida la inamrinona), los síntomas no se aliviaron de manera sistemática, y los inotrópicos dependientes de AMP cíclico se relacionaron de manera sistemática con un aumento del riesgo de hospitalización y muerte. Los pacientes con síntomas de clase IV de la NYHA parecían tener un riesgo particular.
Contraindicaciones
La inamrinona está contraindicada en pacientes hipersensibles a ella.
También está contraindicado en aquellos pacientes que se sabe que son hipersensibles a los bisulfitos.
Advertencias
Contiene metabisulfito de sodio, un sulfito que puede causar reacciones de tipo alérgico, incluidos síntomas anafilácticos y episodios asmáticos menos graves o potencialmente mortales en ciertas personas susceptibles. La prevalencia general de sensibilidad a los sulfitos en la población general es desconocida y probablemente baja. La sensibilidad a los sulfitos se observa con más frecuencia en personas asmáticas que en personas no asmáticas.
Precauciones
General
La inamrinona no debe utilizarse en pacientes con enfermedad valvular aórtica o pulmonar grave en lugar de alivio quirúrgico de la obstrucción. Al igual que otros agentes inotrópicos, puede agravar la obstrucción del tracto de salida en la estenosis subaórtica hipertrófica.
Durante el tratamiento intravenoso con Inamrinona, se debe controlar la presión arterial y la frecuencia cardíaca y disminuir o detener la velocidad de perfusión en pacientes que presenten disminuciones excesivas de la presión arterial.
Los pacientes que han recibido tratamiento diurético vigoroso pueden tener una presión de llenado cardíaca insuficiente para responder adecuadamente a la Inamrinona, en cuyo caso puede estar indicada una liberalización prudente de la ingesta de líquidos y electrolitos.
Se han observado arritmias supraventriculares y ventriculares en la población de muy alto riesgo tratada. Mientras que la inamrinona per se no ha demostrado ser arritmogénica, el potencial de arritmia, presente en la propia insuficiencia cardíaca congestiva, puede aumentar con cualquier medicamento o combinación de medicamentos.
Se han observado trombocitopenia y hepatotoxicidad (ver REACCIONES ADVERSAS).
USO EN EL INFARTO AGUDO DE MIOCARDIO
No se han realizado ensayos clínicos en pacientes en la fase aguda del infarto postmiocárdico. Por lo tanto, la inamrinona no se recomienda en estos casos.
Pruebas de laboratorio
Líquidos y Electrolitos
Durante el tratamiento con Inamrinona se deben monitorizar cuidadosamente los cambios de líquidos y electrolitos y la función renal. La mejoría del gasto cardíaco con la diuresis resultante puede requerir una reducción de la dosis de diurético. La pérdida de potasio debida a una diuresis excesiva puede predisponer a los pacientes digitalizados a arritmias. Por lo tanto, la hipopotasemia debe corregirse con suplementos de potasio antes o durante el uso de inamrinona.
Interacciones medicamentosas
En una experiencia relativamente limitada, no se han observado manifestaciones clínicas adversas en pacientes en los que se utilizó Inamrinona simultáneamente con los siguientes medicamentos: glucósidos digitálicos; lidocaína, quinidina; metoprolol, propranolol; hidralazina, prazosina; dinitrato de isosorbida, nitroglicerina; clortalidona, ácido etacrínico, furosemida, hidroclorotiazida, espironolactona, captopril, heparina, warfarina, suplementos de potasio, insulina, diazepam.
Se ha notificado un caso de hipotensión excesiva cuando se utilizó inamrinona simultáneamente con disopiramida.
Hasta que se disponga de experiencia adicional, la administración concomitante de disopiramida debe realizarse con precaución.
Interacciones químicas
Una interacción química ocurre lentamente durante un período de 24 horas cuando la solución intravenosa de Inamrinona se mezcla directamente con soluciones que contienen dextrosa (glucosa). POR LO TANTO, LA Inamrinona NO DEBE DILUIRSE CON SOLUCIONES QUE CONTENGAN DEXTROSA (GLUCOSA) ANTES DE LA INYECCIÓN.
Se produce inmediatamente una interacción química, que se evidencia por la formación de un precipitado cuando se inyecta furosemida en una vía intravenosa de infusión de Inamrinona. Por lo tanto, la furosemida no debe administrarse en vías intravenosas que contengan inamrinona.
Carcinogénesis, Mutagénesis, Alteración de la fertilidad
No hubo indicios de potencial carcinogénico con Inamrinona cuando se administró por vía oral durante un máximo de dos años a ratas y ratones a niveles de dosis hasta la dosis máxima tolerada de 80 mg/kg/día.
La prueba de micronúcleos de ratón (de 7,5 a 10 veces la dosis máxima en humanos) y la prueba de aberración cromosómica de ovario de hámster chino fueron positivas, lo que indica tanto el potencial clastogénico como la supresión del número de eritrocitos policromáticos. Sin embargo, el ensayo de salmonella de Ames, el estudio de linfoma de ratón y el análisis de metafase de linfocitos humanos cultivados fueron negativos. Los efectos clastogénicos contrastan con los resultados negativos obtenidos en los estudios de fertilidad en ratas macho y hembra, y en un estudio de tres generaciones en ratas, ambos con administración oral.
En estos estudios se observó una ligera prolongación del período de gestación en ratas a niveles de dosis de 50 mg / kg / día y 100 mg/kg/día. La distocia se produjo en madres que recibieron 100 mg / kg / día, lo que resultó en un aumento del número de nacidos muertos, una disminución del tamaño de la camada y una supervivencia deficiente de las crías.
Embarazo
Efectos teratogénicos-Embarazo de categoría C
En conejos blancos de Nueva Zelanda, se ha demostrado que la inamrinona produce malformaciones óseas fetales y externas macroscópicas a dosis orales de 16 mg/kg y 50 mg / kg que eran tóxicas para el conejo. Los estudios en conejos franceses Hy/Cr utilizando dosis orales de hasta 32 mg / kg / día no confirmaron este hallazgo. No se observaron malformaciones en ratas que recibieron inamrinona por vía intravenosa a la dosis máxima utilizada, 15 mg/kg/día (aproximadamente la dosis intravenosa diaria recomendada para pacientes con insuficiencia cardíaca congestiva). No hay estudios adecuados y bien controlados en mujeres embarazadas. La inamrinona se debe usar durante el embarazo solo si el beneficio potencial justifica el riesgo potencial para el feto.
Madres lactantes
Se debe tener precaución cuando se administra Inamrinona a mujeres lactantes, ya que se desconoce si se excreta en la leche materna.
Uso pediátrico
No se ha establecido la seguridad y eficacia en pacientes pediátricos.
Reacciones Adversas
Trombocitopenia: La inyección intravenosa de inamrinona produjo reducciones del recuento de plaquetas por debajo de 100.000/mm3 o límites normales en el 2,4% de los pacientes.
es más común en pacientes que reciben terapia prolongada. Hasta la fecha, en ensayos clínicos estrechamente monitorizados, no se han observado fenómenos hemorrágicos en pacientes cuyos recuentos de plaquetas no se permitió que permanecieran deprimidos.
La reducción de plaquetas depende de la dosis y aparece debido a una disminución en el tiempo de supervivencia plaquetaria. Varios pacientes que desarrollaron trombocitopenia mientras recibían inamrinona se sometieron a exámenes de médula ósea normales. No hay evidencia que relacione la reducción plaquetaria con la respuesta inmunitaria o con un factor activador plaquetario.
Efectos gastrointestinales: Las reacciones adversas gastrointestinales notificadas con Inamrinona durante el uso clínico incluyeron náuseas (1,7%), vómitos (0,9%), dolor abdominal (0,4%) y anorexia (0,4%).
Efectos cardiovasculares: Las reacciones adversas cardiovasculares notificadas con Inamrinona incluyen arritmia (3%) e hipotensión (1,3%).
Toxicidad hepática: En perros, a dosis intravenosas de entre 9 mg / kg / día y 32 mg/kg/día, la inamrinona mostró hepatotoxicidad relacionada con la dosis, manifestada como elevación enzimática o necrosis de células hepáticas, o ambas. Se ha observado hepatotoxicidad en el hombre tras la administración oral a largo plazo y, en una experiencia limitada (0,2%), tras la administración intravenosa de inamrinona. También se han notificado casos raros de elevación de enzimas y bilirrubinas e ictericia.Hipersensibilidad: Se han notificado varias reacciones de hipersensibilidad aparentes en pacientes tratados con Inamrinona oral durante aproximadamente dos semanas. Los signos y síntomas fueron variables, pero incluyeron pericarditis, pleuritis y ascitis (1 caso), miositis con sombra intersticial en la radiografía de tórax y tasa de sedimentación elevada (1 caso) y vasculitis con densidades pulmonares nodulares, hipoxemia e ictericia (1 caso). El primer paciente murió, no necesariamente de la posible reacción, mientras que los dos últimos se resolvieron con la interrupción del tratamiento. Ninguno de los casos se volvió a plantear, por lo que la atribución a la inamrinona no es segura, pero se deben considerar posibles reacciones de hipersensibilidad en cualquier paciente mantenido durante un período prolongado con inamrinona.
General: Las reacciones adversas adicionales observadas en los ensayos clínicos con Inamrinona intravenosa incluyen fiebre (0,9%), dolor en el pecho (0,2%) y ardor en el lugar de la inyección (0,2%).
Manejo de las Reacciones Adversas
Reducción del Recuento de Plaquetas: La reducción asintomática del recuento de plaquetas (a <150.000/mm3) puede revertirse en el plazo de una semana a partir de la disminución de la dosis del medicamento. Además, sin cambios en la dosis del fármaco, el recuento puede estabilizarse a niveles inferiores a los previos al fármaco sin secuelas clínicas. Se recomiendan recuentos plaquetarios previos al fármaco y recuentos plaquetarios frecuentes durante el tratamiento para ayudar a tomar decisiones con respecto a las modificaciones de dosis.
Si se produce un recuento de plaquetas inferior a 150.000 / mm3, se pueden considerar las siguientes acciones:
* Mantener la dosis diaria total sin cambios, ya que en algunos casos los recuentos se han estabilizado o han vuelto a los niveles previos al tratamiento. * Disminuir la dosis diaria total. * Suspender la administración de Inamrinona si, a juicio clínico del médico, el riesgo supera el beneficio potencial.
Efectos secundarios gastrointestinales: Si bien los efectos secundarios gastrointestinales se observaron con poca frecuencia con la terapia intravenosa, en caso de que ocurrieran efectos graves o debilitantes, el médico puede desear reducir la dosis o suspender el medicamento según las consideraciones habituales de beneficio a riesgo.
Toxicidad hepática: En la experiencia clínica hasta la fecha con la administración intravenosa, se ha observado hepatotoxicidad en raras ocasiones. Si se producen alteraciones agudas marcadas de las enzimas hepáticas junto con síntomas clínicos que sugieran una reacción de hipersensibilidad idiosincrásica, se debe interrumpir inmediatamente el tratamiento con inamrinona.
Si se producen alteraciones enzimáticas menos que marcadas sin síntomas clínicos, estos cambios inespecíficos deben evaluarse de forma individual. Es posible que el médico desee continuar con la inamrinona, reducir la dosis o suspender el medicamento según las consideraciones usuales de beneficio/riesgo.
Sobredosis
Se ha notificado una muerte con una sobredosis masiva accidental (840 mg durante tres horas por bolo inicial y perfusión) de Inamrinona, aunque la relación causal es incierta. Se debe actuar con diligencia durante la preparación y administración del producto.
Las dosis de inamrinona pueden producir hipotensión debido a su efecto vasodilatador. Si esto ocurre, se debe reducir o suspender la administración de inamrinona. No se conoce ningún antídoto específico, pero se deben tomar medidas generales para el soporte circulatorio.
En ratas, la LD50 de Inamrinona, como sal de lactato, fue de 102 mg/kg o 130 mg/kg por vía intravenosa en dos estudios diferentes y de 132 mg/kg por vía oral (intragástrica); como suspensión en tragacanto de goma acuosa, la LD50 oral fue de 239 mg/kg.
Dosis y administración de Inamrinona
Las dosis de carga de la inyección de Inamrinona deben administrarse según se suministran (sin diluir). Las perfusiones de inamrinona pueden administrarse en solución salina normal o media normal a una concentración de 1 mg/ml a 3 mg/ml. Las soluciones diluidas deben utilizarse en un plazo de 24 horas.
La inyección de inamrinona se puede administrar en perfusiones de dextrosa (glucosa) a través de un conector en Y o directamente en el tubo cuando sea preferible.
Interacciones químicas
Una interacción química ocurre lentamente durante un período de 24 horas cuando la solución intravenosa de Inamrinona se mezcla directamente con soluciones que contienen dextrosa (glucosa). POR LO TANTO, LA Inamrinona NO DEBE DILUIRSE CON SOLUCIONES QUE CONTENGAN DEXTROSA (GLUCOSA) ANTES DE LA INYECCIÓN.
Se produce inmediatamente una interacción química, que se evidencia por la formación de un precipitado cuando se inyecta furosemida en una vía intravenosa de infusión de Inamrinona. Por lo tanto, la furosemida no debe administrarse en vías intravenosas que contengan inamrinona.
Se recomienda el siguiente procedimiento para la administración de la inyección de inamrinona:
1. Inicie el tratamiento con una dosis de carga de 0,75 mg/kg administrada lentamente durante 2 a 3 minutos.
|
LOADING DOSE DETERMINATION 0.75 mg/kg (undiluted) |
||||||||||
|
Patient Weight in kg |
||||||||||
|
mL of undiluted Inamrinone Injection |
||||||||||
2. Continue therapy with a maintenance infusion between 5 mcg/kg/min and 10 mcg/kg/min.
3. Based on clinical response, an additional loading dose of 0.75 mg/kg may be given 30 minutes after the initiation of therapy.
4. La velocidad de perfusión suele oscilar entre 5 mcg / kg / min y 10 mcg/kg/min, de modo que la dosis diaria total recomendada (incluidas las dosis de carga) no supera los 10 mg / kg. Un número limitado de pacientes estudiados a dosis más altas apoyan un régimen de dosificación de hasta 18 mg/kg/día para duraciones de tratamiento más cortas.
Se puede utilizar la siguiente tabla de velocidad de perfusión para garantizar que los cálculos se realizan correctamente.
Para utilizar la tabla, la concentración de la solución de infusión de inamrinona utilizada debe ser de 2,5 mg/ml (2500 mcg / mL). Esta concentración se prepara mezclando la solución de inamrinona con un volumen igual de diluyente (solución salina normal o media normal).
| * Dilución: Para preparar la concentración de 2,5 mg/ml recomendada para perfusión, mezcle Inamrinona con un volumen igual de diluyente. Por ejemplo, mezclar tres viales de 20 ml de Inamrinona (3 x 20 ml = 60 ml) con 60 ml de diluyente para obtener un volumen total de 120 mL de la solución final de 2,5 mg/ml de Inamrinona. | ||||||||||
|
Inamrinone IV INFUSION RATE (mL/hr) CHART Using 2.5 mg/mL Infusion Concentration* |
||||||||||
|
Patient Weight in kg |
||||||||||
|
Dosage: 5.0 mcg/kg/min |
||||||||||
|
7.5 mcg/kg/min |
||||||||||
|
10.0 mcg/kg/min |
||||||||||
Example: A 70 kg patient would require a loading dose of 10.5 mL of undiluted Inamrinone. If the physician selects a dose of 7.5 mcg / kg / min para la perfusión, el caudal sería de 13 ml / h a la concentración de 2,5 mg/ml de Inamrinona.
5. La velocidad de administración y la duración del tratamiento deben ajustarse en función de la respuesta del paciente. El médico puede querer reducir o ajustar la perfusión a la baja en función de la respuesta clínica o de los efectos adversos.
Se puede esperar que los regímenes de dosificación anteriores sitúen la concentración plasmática de Inamrinona de la mayoría de los pacientes en aproximadamente 3 mcg / ml. Los aumentos en el índice cardíaco muestran una relación lineal con la concentración plasmática de un rango de 0.5 mcg/ml a 7 mcg/ml. No se han realizado observaciones a concentraciones plasmáticas mayores.
La mejoría del paciente puede reflejarse en aumentos del gasto cardíaco, reducción de la presión de cuña capilar pulmonar y respuestas clínicas tales como una disminución de la disnea y una mejoría de otros síntomas de insuficiencia cardíaca, como la ortopnea y la fatiga.
La monitorización de la presión venosa central (PVC) puede ser valiosa en la evaluación de la hipotensión y el manejo del equilibrio de líquidos. La corrección o ajuste previo de líquidos / electrolitos es esencial para obtener una respuesta satisfactoria con Inamrinona.
Los medicamentos parenterales deben inspeccionarse visualmente y no deben utilizarse si se observan partículas o decoloración.
Cómo se suministra la Inamrinona
La inyección de inamrinona USP se suministra en viales monodosis de 20 ml de solución estéril de color amarillo transparente en caja individual. NDC 55390-042-10.
Cada 1 ml contiene lactato de inamrinona equivalente a 5 mg de Inamrinona.
Proteger de la luz. El embalaje es resistente a la luz para protegerlo durante el almacenamiento. Conservar en el envase hasta el momento de su uso.
Almacenar a temperatura ambiente controlada de 15° a 30° C (59°a 86 ° F).
Manufactured by: Manufactured for:
Ben Venue Laboratories, Inc. Bedford Laboratories™
Bedford, OH 44146 Bedford, OH 44146
August 2002 AMR-P01
VIAL LABEL

Vial Label 100 mg/20 mL
| Inamrinone Inamrinone injection |
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
Labeler – Bedford Laboratories (884528407)
Registrant – Ben Venue Laboratories (004327953)
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Ben Venue Laboratories Inc. | 004327953 | MANUFACTURE(55390-042) | |
More about inamrinone
- Side Effects
- During Pregnancy
- Dosage Information
- Drug Interactions
- Drug class: inotropic agents
Related treatment guides
- Heart Failure
Medical Disclaimer