Los aminoácidos, nucleótidos o cualquier otro carácter evolutivo son reemplazados por otros a cierta velocidad. Por ejemplo, imagine una secuencia evolutiva con threepossible los estados, a, B y C. Si el modelo de sustitución de tiempo es reversible, therewill ser tres las tasas de transición, de Un<>B, B<>C y Un<>C.
Supongamos que las tasas son 1, 1 y 0 respectivamente en unidades de sustitución por 100 caracteres por unidad de tiempo. Después de una unidad de tiempo, en una secuencia de 300 caracteres compuesta originalmente por igual de As, Bs y Cs, esperamos que haya habido una sustitución de A a B y una sustitución de B a C. Si comparamos dos secuencias homólogas en organismos vivos, porque se ha superado una unidad de tiempo para ambas secuencias, esperaríamos dos A a B y dos B a Csubstituciones entre las secuencias actuales.
No importa durante cuánto tiempo ejecutemos este proceso, nunca habrá una sustitución directa de A por C. Tampoco habrá una sustitución de A a C bajo el modelo de sitios infinitos llamado aso, donde no se puede producir más de una sustitución en un solo sitio.
Sin embargo, dado que las sustituciones de A a B y de B a C son comunes, bajo un modelo de sitios finitos, eventualmente, B será reemplazado por C en un sitio donde A fue reemplazada previamente por B. Este reemplazo indirecto de A por C (o de manera equivalente en un modelo reversible a tiempo, C por A) se vuelve más probable cuanto más largo sea el período de separación de las secuencias homólogas.
Simulé la evolución de la secuencia basada en el escenario anterior, ejecutando la simulación durante 10 unidades de tiempo. A partir de esta sustitución, observé los siguientes recuentos para cada patrón de sitio:
| A | B | C | |
|---|---|---|---|
| A | 91 | 9 | 0 |
| B | 5 | 86 | 9 |
| C | 0 | 9 | 91 |
Dentro de este relativamente de corta duración, no parece como si cualquier de Un<>Csubstitutions se han producido. Sin embargo, cuando vuelvo a escanear la simulación por 100 unidades de tiempo:
| A | B | C | |
|---|---|---|---|
| A | 55 | 35 | 10 |
| B | 29 | 36 | 35 |
| C | 20 | 36 | 44 |
Como se puede ver, muchos «A» personajes han sido reemplazados con «C» y viceversa. De manera más general, bajo un modelo de sitios finitos, las sustituciones múltiples hacen que la distribución de los recuentos de patrones de sitios se vuelva mucho más plana más allá de aumentar la proporción de conteos diagonales en relación con los diagonales.Las matrices de puntuación PAM y BLOSUM tienen en cuenta múltiples sustituciones de diferentes maneras.
Las matrices PAM para aminoácidos, junto con las abreviaturas de una sola letra utilizadas para los aminoácidos codificados genéticamente, fueron desarrolladas por MargaretDayhoff. Se publicaron originalmente en 1978, y se basaron en las secuencias de proteínas que Dayhoff había estado compilando desde la década de 1960, publicadas como theAtlas of Protein Sequence and Structure.
El nombre PAM proviene de» mutación aceptada en el punto», y se refiere a la sustitución de un solo aminoácido en una proteína con un aminoácido diferente.Estas mutaciones se identificaron comparando secuencias muy similares con al menos un 85% de identidad, y se supone que cualquier sustitución observada fue el resultado de una única mutación entre la secuencia ancestral y una de las secuencias actuales del día.
PAM también define una unidad de tiempo, donde 1 PAM es el tiempo en el que se espera que 1/100 aminoácidos experimenten una mutación. La matriz de probabilidad PAM1 muestra la probabilidad de que el aminoácido en la columna j sea reemplazado por el aminoácido en la fila i. Se calculó a partir de los recuentos PAM de Dayhoff, y se reajustó a 1 unidad de tiempo PAM. Como puede ver, las probabilidades fuera de la diagonal en la matriz PAM1 son todas muy pequeñas (todos los elementos se escalaron en 10.000 para la legalidad):
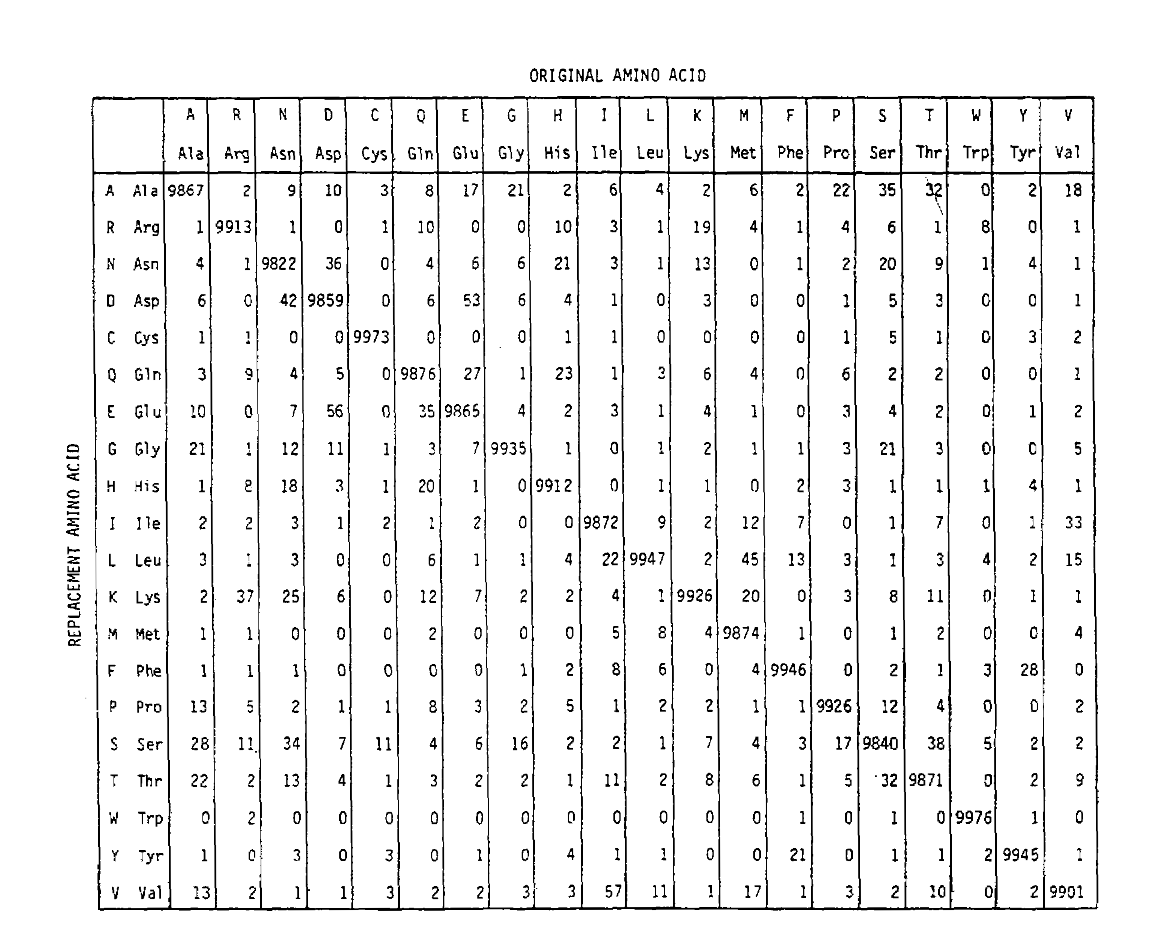
Para calcular las probabilidades de reemplazo de aminoácidos para duraciones de tiempo más largas, la matriz se puede multiplicar por sí misma el número de veces correspondiente. Por lo tanto, la matriz de probabilidad PAM250, que describe las probabilidades de recolocación dadas 250 unidades de tiempo PAM, se derivó elevando la matriz de probabilidad PAM1 a la potencia 250 (todos los elementos se escalaron en 100 para legibilidad):
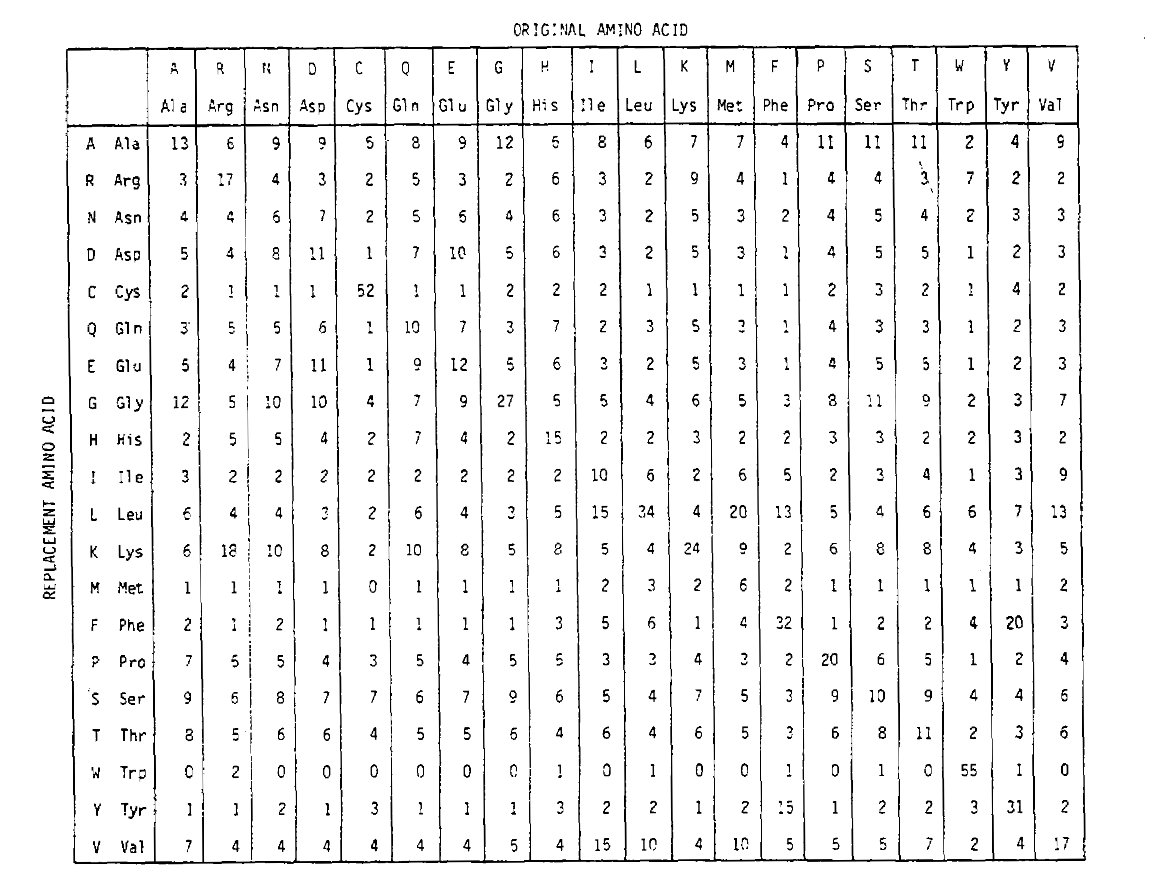
Las probabilidades de reemplazo derivadas utilizando esta exponenciación correctamente cuentan para múltiples sustituciones. No solo las probabilidades no diagonales son proporcionalmente mayores como cabría esperar para una duración de tiempo más larga, sino que son más planas. Por ejemplo, la probabilidad de un reemplazo de valina (V)a isoleucina (I) es 33× mayor que un reemplazo de V a histadina (H)en la matriz PAM1, pero solo 4,5× mayor en la matriz PAM250.
Las matrices de puntuación se pueden calcular a partir de las matrices de probabilidad y las frecuencias base observadas.
Las matrices BLOSUM, desarrolladas por Steven y Jorja Henikoff y publicadas en 1992, tienen un enfoque muy diferente. Mientras que PAM está aplicando implícitamente un modelo de evolución de sitios finitos estacionarios utilizando la exponenciación de matrices, el efecto de sustituciones múltiples se trata implícitamente en BLOSUM mediante la construcción de diferentes matrices de puntuación para diferentes escalas de tiempo.
Dentro de múltiples alineaciones de secuencias homólogas, se identifican bloques contiguos conservados de aminoácidos. Dentro de cada bloque, las secuencias múltiples se agrupan cuando su identidad de secuencia media en pares es superior a algún umbral. El umbral es del 80% para la matriz BLOSUM80, del 62% para BLOSUM62, del 50% para BLOSUM50 y así sucesivamente.
Esto significa que para BLOSUM80, los bloques tendrán una identidad media de pares no superior al 80%, para BLOSUM62 no superior al 62%, etc.
Las probabilidades de reemplazo de aminoácidos para secuencias homólogas se calculan a partir de comparaciones de pares entre grupos. Estas probabilidades serán el resultado de sustituciones simples y múltiples, con sustituciones múltiples que tendrán una mayor influencia a distancias evolutivas mayores. Por lo tanto, las puntuaciones generadas a partir de comparaciones en pares entre grupos de mayor distancia media, como la matriz BLOSUM50, explicarán naturalmente el efecto más amplio de las sustituciones múltiples.
Aunque toman rutas diferentes, las matrículas de puntuación BLOSUM y PAM finales son en realidad bastante similares. Según Henikoff y Henikoff, las siguientes matrices Pam y BLOSUM son comparables:
| PAM | BLOSUM |
|---|---|
| PAM250 | BLOSUM45 |
| PAM160 | BLOSUM62 |
| PAM120 | BLOSUM80 |
For more information on PAM (Dayhoff) and BLOSUM matrices, see capítulo 2 de Análisis de secuencias biológicas por Durbin et al. y Wikipedia.
Actualización del 13 de octubre de 2019: para obtener otra perspectiva sobre las matrices de sustitución, consulte la sección «Desvíos» al final del Capítulo 5 de Algoritmos Bioinformáticos (2a o 3a Edición) de Compeau y Pevzner.