ennek az oktatóanyagnak az a célja, hogy megismertesse Önt a következő generációs szekvenálási adatok feldolgozásával a galaxisban. Ez az oktatóanyag COVID-19 variánshívást használ az Illumina data-ból, de önmagában nem a variánshívásról szól.
a befejezése ez a bemutató tudni fogja:
- hogyan lehet megtalálni az adatokat SRA és át ezt az információt Galaxy
- hogyan kell elvégezni az alapvető NGS adatfeldolgozás Galaxy beleértve:
- minőség-ellenőrzés (QC) Illumina adatok
- Mapping
- eltávolítása másolatok
- Variant hívás
lofreq - Variant annotation
- használata adatkészletek gyűjtemények
- Adatok importálása Jupyter
### napirend>> ebben az oktatóanyagban fogjuk fedezni:>> 1. TOC> {: toc}> {: .napirend} # # két út ezen az oktatóanyagon keresztülkét pályát hoztunk létre, amelyeket ezen az oktatóanyagon keresztül követhet.1. ** 1. pálya * * -kezdje az NCBI SRA-jával, és keresse meg a rendelkezésre álló hozzáféréseket (#the-sequence-read-archive)2. ** Trajectory 2 * * – bypass NCBI SRA és indítsa el a Galaxy közvetlenül. 6. rajt (#Back-in-galaxy) azt javasoljuk, hogy kezdje a **2.pályával**.# A sequence Read ArchiveThe (https://www.ncbi.nlm.nih.gov/sra) a *nem összeszerelt olvasmányok* elsődleges archívuma a (https://www.ncbi.nlm.nih.gov/) számára. Az SRA remek hely a publikációk és tanulmányok alapjául szolgáló szekvenálási adatok megszerzésére.Ez az oktatóanyag bemutatja, hogyan lehet szekvenciaadatokat szerezni az SRA-ból a galaxisba a kettő közötti közvetlen kapcsolat segítségével.> ### comment Comment>> hallani fogja az SRA-t is, amelyet *rövid olvasási archívumnak* neveznek, eredeti nevét.> {:.megjegyzés} # # a SRASRA elérése közvetlenül a weboldalán keresztül, vagy a Galaxy eszközpaneljén keresztül érhető el.> ### comment Comment>> kezdetben az SRA eléréséhez szükséges eszközpanel opció csak a (https://usegalaxy.org/). Az SRA-val való közvetlen kapcsolat támogatását a Galaxy {: 20.05 kiadása tartalmazza .comment}> ### hands_on Hands-on: fedezze fel az SRA Entrez-t>> 1. Menjen a választott galaxis példányához, például az egyik (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) vagy bármely más. (Ez a bemutató használ usegalaxy.org).> 1. Ha az előzmények még nem üresek, akkor indítson új előzményeket (lásd (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) A galaxis történeteiről)> 1. ** Kattintson* * `Adatok beolvasása ‘ az eszközpanel tetején.> 1. ** Kattintson * * ‘SRA Server’ az eszközök listája alatt látható ‘Get Data’.>ez viszi a (https://www.ncbi.nlm.nih.gov/sra) — akkor is indul közvetlenül a SRA. Az oldal tetején megjelenik egy keresőmező. Próbáljon meg keresni valamit, ami érdekli, például ‘delfin’ vagy ‘vese `vagy` delfin vese`, majd **kattintson** a` Keresés’ gombra.>> ez a *sra kísérletek* listáját adja vissza, amelyek megfelelnek a keresési karakterláncnak. Az SRA kísérletek, más néven* SRX bejegyzések*, tartalmaznak egy adott kísérlet szekvenciaadatait, valamint magának a kísérletnek és minden más kapcsolódó adatnak a magyarázatát. A visszaküldött kísérleteket a nevükre kattintva fedezheti fel. Lásd (https://www.ncbi.nlm.nih.gov/könyvek/NBK56913/) a (https://www.ncbi.nlm.nih.gov/ könyvek/n/helpsrakb/) további.>>amikor szöveget ír be az SRA keresőmezőbe, akkor azt használja (https://www.ncbi.nlm.nih.gov/sra /docs/srasearch/). Az Entrez támogatja mind az egyszerű szöveges kereséseket, mind a nagyon pontos kereséseket, amelyek bizonyos metaadatokat ellenőriznek és önkényesen összetett logikai kifejezéseket használnak. Az Entrez lehetővé teszi, hogy a kereséseket az alaptól a haladóig növelje, miközben szűkíti a kereséseket. A speciális keresések szintaxisa ijesztőnek tűnhet, de az SRA grafikus (https://www.ncbi.nlm.nih.gov/sra/advanced/) az adott szintaxis létrehozásához. Az sra Run Selector még barátságosabb felhasználói felületet biztosít a kiválasztott adatok szűkítéséhez.>> játsszon körül az SRA Entrez interfésszel, beleértve az advanced query builder-t is, hogy megtudja, azonosíthat-e olyan SRA kísérleteket, amelyek relevánsak az egyik kutatási területe szempontjából.{: .hands_on}> # # # hands_on Hands-on: Generálja a megfelelő kísérletek listáját az Entrez használatával>> most, hogy alapszintű ismerete van az SRA Entrez-nek, keressük meg az ebben az oktatóanyagban használt szekvenciákat.>> 1. Ha még nem vagy ott, * * navigáljon * * vissza a (https://www.ncbi.nlm.nih.gov/sra> 1. ** Tiszta* * minden keresési szöveget a keresőmezőbe.> 1. ** Írja be* * ‘ sars-cov-2` a keresőmezőbe, majd **Kattintson** `Keresés`.> ez az SRA kísérletek hosszú listáját adja vissza, amelyek megfelelnek a keresésünknek, és ez a lista túl hosszú ahhoz, hogy egy oktatógyakorlatban használhassuk. Ezen a ponton tudtuk használni a fejlett Entrez query builder tanultunk fent.> de nem fogjuk. ehelyett küldjük el a *too long for a tutorial* list találatokat az SRA Run választóhoz, és használjuk a barátságosabb felületét az eredmények szűkítéséhez.>> !(../../ képek / sra_entrez.png) {:.hands_on}> # # # hands_on Hands-on: Ugrás az Entrez-ről az SRA Futásválasztóra>> az eredményeket kibővített interaktív táblaként tekintheti meg a RunSelector segítségével.>> 1. Kattintson a gombra eredmények küldése a futtatáshoz választó, amely a keresési eredmények tetején található mezőben jelenik meg.>> !(../../ képek / sra_entrez_result.png)>>> ### tipp mi van, ha nem látja a Futtatás választó linket?>>>>> lehet, hogy korábban észrevette ezt a szöveget, amikor az Entrez keresést kutatta. Ez a szöveg csak néhány alkalommal jelenik meg, amikor a keresési eredmények száma meglehetősen széles ablakba esik. Nem fogja látni, ha csak néhány eredménye van, és nem fogja látni, ha több eredménye van, mint amennyit a Futtatás választó el tud fogadni.>>>>> *be kell, hogy fut Selector küldeni az eredményeket a Galaxy.* Mi van, ha nincs elég eredménye a link megjelenítéséhez? Ebben az esetben az eredménypanel jobb felső sarkában található `Küldés` legördülő menüre kattintva *** a get to run választót hívja. A Futásválasztó eléréséhez** válassza a * * `Futásválasztó ‘ lehetőséget, majd** kattintson * * a `Go` gombra.>!(../../ képek / sra_entrez_send_to.png) > {: .tipp}>>> 1. ** Kattintson * * ‘Send results to Run selector’ a tetején a keresési eredmények panel. (Ha nem látja ezt a linket, akkor nézze meg közvetlenül a fenti megjegyzést.){: .hands_on} # # SRA Run Selectorkorábban megtanultuk, hogyan szűkíthetjük Keresési eredményeinket az Entrez speciális szintaxisával. Ezt az erőt azonban nem használtuk ki, amikor Entrezben voltunk. Ehelyett egy egyszerű keresést használtunk, majd az összes eredményt elküldtük a Futásválasztónak. Még nincs meg az eredmények (rövid) listája, amelyen elemzést szeretnénk futtatni. * Mit csinálunk?* Mi használ Entrez és a Run Selector hogyan tervezték, hogy kell használni: * Használja a Entrez felület szűkíteni az eredményeket le a méretet, hogy a Run Selector fogyaszthat. * Küldje el ezeket Entrez eredményeket az SRA Run Selector * használja a Run Selector sokkal barátságosabb felület 1. Könnyebb megérteni a rendelkezésünkre álló adatokat 1. Szűkítse ezeket az eredményeket a tudás felhasználásával.> # # # comment A Futásválasztó több és kevesebb, mint az Entrez>> A Futásválasztó a legtöbb, de nem minden, amit az Entrez Keresési szintaxisa képes. Run selector használ * csiszolt keresés * technológia, amely könnyen használható, és nagy teljesítményű, de amely velejárója korlátokat. Pontosabban, az Entrez jobban fog működni, ha olyan attribútumokra keres, amelyek több tíz, száz vagy ezer különböző értékkel rendelkeznek. Run Selector jobban fog működni kereső attribútumok kevesebb, mint 20 különböző értékeket. Szerencsére ez leírja a legtöbb keresést.{: .megjegyzés}a Futtatás választó ablak több panelre oszlik: * * * `szűrők listája’**: a bal felső sarokban. Ez az, ahol finomítani fogjuk a keresést.* * * ‘Select’**: összefoglaló arról, hogy mi volt eredetileg át a Run Selector, és hogy mennyi, hogy már kiválasztott eddig. (Eddig még nem választottunk egyet sem.) Vegye figyelembe a kínzó, de még mindig szürkén megjelenő `Galaxy ‘ gombot is.** * ` Talált x elemek ‘ * * kezdetben ez az entrez által a Futásválasztóhoz küldött elemek listája. Ez a lista zsugorodik, amikor szűrőket alkalmazunk rá.!(../../ képek / sra_run_selector.png)> ### megjegyzés miért emelkedett a talált elemek száma*?* >> emlékezzünk arra, hogy az Entrez felület felsorolja az SRA kísérleteket (SRX bejegyzések). Run Selector listák * fut* – szekvenálás adatkészletek — és vannak* egy vagy több * fut per kísérlet. Ugyanazok az adatok vannak, mint korábban, most csak finomabb részletekben látjuk.{: .megjegyzés}a bal felső sarokban található `szűrők listája ‘ olyan oszlopokat jelenít meg az eredményeinkben, amelyek folyamatos numerikus értékekkel rendelkeznek, vagy 10 vagy annál kevesebb (ezt a számot megváltoztathatja) különálló értékekkel rendelkeznek. ** Görgessen* * lefelé a listán válasszon ki néhány szűrőt. Ha egy szűrő van kiválasztva, az alábbiakban megjelenik egy *értékek* mező, amely felsorolja a szűrő beállításait, valamint az egyes opciókhoz tartozó futtatások számát. Ezek az értékek / opciók az adatkészlet metaadataiból származnak. Próbálja * * kiválasztása * * néhány érdekes hangzó szűrőket, majd * * válasszon * * egy vagy több lehetőséget minden szűrő. Próbálja meg * * kijelölés megszüntetése * * beállítások és szűrők. Ennek során a talált eredmények száma csökken vagy növekszik.> # # # tip Tip: Használjon szűrőket az adatok jobb megértéséhez>> A szűrők segítségével szűkítheti a vizsgált adatkészleteket a Galaxy-ba történő küldéshez, de kiváló módja az adatok megértésének is:> először is, a szűrő kiválasztásával egyszerűen megtekintheti az oszlop értéktartományát. Lehet, hogy nem tudja (https://www.google.com/search?q=sra+sirs_outcome), de valószínűleg kitalálhatja, hogy milyen értékek vannak benne.> másodszor felfedezheti, hogy a különböző oszlopok hogyan kapcsolódnak egymáshoz. Van-e összefüggés a `sirs_outcome` értékek és a `disease_stage` értékek között?{: .tipp}> ### hands_on Hands-on: szűkítse az eredményeket a Futásválasztó segítségével>> 1. Ha bármilyen szűrő be van kapcsolva, **törölje a kijelölést** őket.> miután ezt megtette, nem jelennek meg *értékek* dobozok a `szűrők listája`alatt.> 2. ** Másolja be* * ezt a keresési karakterláncot a ‘talált elemek’ keresőmezőbe.>> SRR11772204 OR SRR11597145 OR SRR11667145>> ez a kéz-felvette sor fut korlátozza az eredményeket 3 fut a különböző földrajzi eloszlás.{: .hands_on}ez csökkenti a` talált elemek ‘ listáját több tízezer futásról 3 futásra (kezelhető szám egy oktatóanyaghoz!). De még nem végeztünk a Futásválasztóval. Vegye figyelembe, hogy a` Galaxy ‘ gomb még mindig szürkén jelenik meg. Leszűkítettük a lehetőségeinket, de még nem választottunk ki semmit, amit Elküldhetnénk a Galaxynak.Lehetőség van, hogy kiválassza az összes fennmaradó távon * * kattintva * * a pipa tetején az első oszlop. Mindent törölhet, ha * * rákattint* * az `X` – re.> ### hands_on Hands-on: Válassza ki a futásokat és küldje el a galaxisnak>> 1. Válassza ki az összes futást** az `X`gombra kattintva.> és most a `Galaxy` gomb él.> 1. ** Kattintson* * a `Galaxy ‘gombra a` Select’ részben az oldal tetején.{: .hands_on} # # vissza GalaxyWhen kattintunk ‘ Galaxy` a Futásválasztóban több dolog történik. Először elindít egy új böngésző fület vagy ablakot, amely a Galaxy-ban nyílik meg. Látni fogja a *nagy zöld dobozt*, amely jelzi, hogy az SRA és a Galaxy közötti kézfogás sikeres volt, majd egy új `SRA` munkát fog látni az Előzmények panelen. Ez a mező szürke / függőben lehet, jelezve, hogy az átvitel még nem kezdődött el, vagy egyenesen sárga / futó vagy zöld / kész.> # # # hands_on Hands-on: Vizsgálja meg az új SRA adatkészletet>> 1. Miután az` SRA ‘ átvitel befejeződött, * * kattintson * * az adatkészlet galaxy-eye (eye) ikonjára.>> Ez megjeleníti az adatkészletet a Galaxy középső paneljén.{: .hands_on}az ‘ SRA ‘ adatkészlet nem szekvenciaadatok ,hanem inkább * metaadatok* , amelyeket az sra szekvenciaadatainak lekérésére használunk. Ez a metaadat tükrözi azokat az információkat, amelyeket a Futásválasztó `talált elemek` szakaszában láttunk. A metaadatok nem azok a végadatok, amelyeket az SRA-tól keresünk, de az összes metaadat megléte gyakran hasznos a későbbi elemzési lépésekben.Lehetővé teszi, hogy most használja, hogy a metaadatok letölteni a szekvencia adatokat SRA. Az SRA eszközöket biztosít mindenféle információ kinyerésére, beleértve magát a szekvenciaadatokat is. A Galaxy Tool `Faster Download and Extract Reads in FASTQ` az SRA (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump) segédprogramon alapul, és éppen ezt teszi.– >
- keresse meg a szükséges adatokat az SRA-ban
- hands_on Hands-on: feladat leírása
- megjegyzés Megjegyzés
- folyamat és szűrő SraRunInfo.csv fájl a galaxisban
- hands_on Hands-on: SraRunInfo feltöltése.csv fájlt Galaxy
- megjegyzés Óvakodj a vágásoktól
- hands_on Hands-on: adatok részhalmazának létrehozása
- tip tipp: Eszközök keresése
- töltse le a szekvenálási adatokat a gyorsabb letöltéssel és a kivonat beolvasásával a FASTQ-ban
- hands_on Hands-on: feladat leírása
- most mi van?
- a SARS-Cov-2 szekvenálási adatok variációs elemzése
- megjegyzés a usegalaxy.* COVID-19 elemzési projekt
- Szerezd meg a referencia genomadatokat
- hands_on Hands-on: Szerezd meg a referencia Genom adatait
- tipp: Importálás linkeken keresztül
- Adaptervágás fastp-vel
- hands_on Hands-on: feladat leírása
- igazítás a térképhez a BWA-mem segítségével
- hands_on Hands-on: Igazítsa a szekvenálást a referencia genomhoz
- távolítsa el a másolatokat a MarkDuplicates segítségével
- hands_on Hands-on: távolítsa el a PCR másolatokat
- igazítási statisztikák létrehozása a samtools statisztikákkal
- hands_on Hands-on: Igazítási statisztikák létrehozása
- A lofreq viterbi-vel történő olvasás átrendezése
- hands_on Hands-on: átrendez az indels körül
- adjon hozzá indel tulajdonságokat a lofreq insert indel tulajdonságokkal
- hands_on Hands-on: Add indel adottságok
- hívási változatok a lofreq hívásváltozatok használata
- hands_on Hands-on: Hívásváltozatok
- jegyezze fel a variáns effektusokat az SnpEff eff segítségével:
- hands_on Hands-on: jegyzetekkel variáns hatások
- változótáblázat létrehozása SnpSift Kivonatmezők használatával
- hands_on Hands-on: a változatok táblázatának létrehozása
- összegezze az adatokat a MultiQC-vel
- hands_on Hands-on: az adatok összefoglalása
- következtetés
- kulcspontok kulcsfontosságú pontok
- Gyakran Ismételt Kérdések
- hasznos irodalom
- visszajelzés
- idézve ezt az oktatóanyagot
- details BibTeX
keresse meg a szükséges adatokat az SRA-ban
először meg kell találnunk egy jó adatkészletet, amellyel játszhatunk. A sequence Read Archive (sra) az Egyesült Államok Nemzeti Egészségügyi Intézete (NIH) által működtetett össze nem állított olvasmányok elsődleges archívuma. Az SRA remek hely a publikációk és tanulmányok alapjául szolgáló szekvenálási adatok megszerzésére. Tegyük ezt meg:
hands_on Hands-on: feladat leírása
- LÉPJEN az NCBI SRA oldalára a böngésző https://www.ncbi.nlm.nih.gov/sra
- a keresőmezőbe írja be
SARS-CoV-2 Patient Sequencing From Partners / MGH(Alternatív megoldásként egyszerűen kattintson erre a linkre)
- a weboldal nagyszámú SRA adatkészletet mutat (az írás idején 2223 volt). Ez egy tanulmány adatai, amelyek leírják a SARS-CoV-2 elemzését Boston területén.
- töltse le az adatkészleteket leíró metaadatokat:
- a Küldés: legördülő menüre kattintva
- a
File- formátum megváltoztatása
RunInfo- a fájl létrehozása gombra kattintva így kell kinéznie:
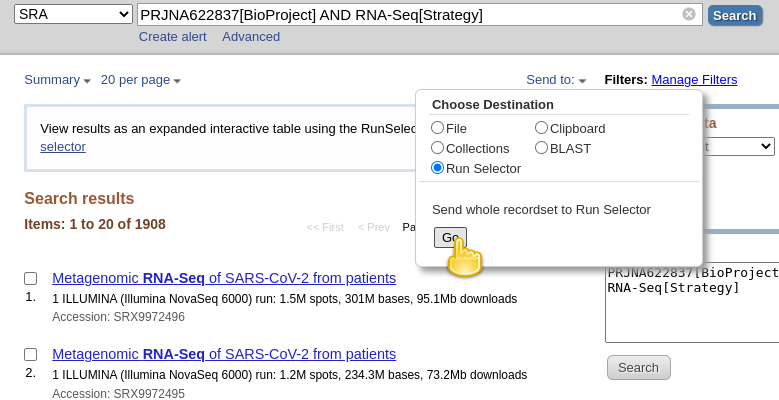
- ez egy meglehetősen nagy
SraRunInfo.csvfájlt hozna létre aDownloadsmappában.
most, hogy letöltöttük ezt a fájlt, elmehetünk egy Galaxy példányba, és elkezdhetjük feldolgozni.
megjegyzés Megjegyzés
vegye figyelembe, hogy az imént letöltött fájl nem maga az adat szekvenálása. Inkább metaadatok, amelyek leírják a szekvenálási olvasások tulajdonságait. Ezt a listát csak néhány csatlakozásra szűrjük le, amelyeket a bemutató hátralévő részében használunk.
folyamat és szűrő SraRunInfo.csv fájl a galaxisban
hands_on Hands-on: SraRunInfo feltöltése.csv fájlt Galaxy
- menj a Galaxy példány választás, mint az egyik usegalaxy.org, usegalaxy.eu, usegalaxy.org.au vagy bármely más. (Ez a bemutató használ usegalaxy.org).
- kattintson az adatok feltöltése gombra:
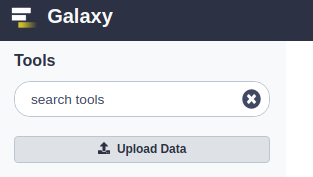
- a megjelenő párbeszédpanelen kattintson a “helyi fájlok kiválasztása” gombra:

- keresse meg és válassza ki a
SraRunInfo.csvfájlt a számítógépről- kattintson a Start gombra
- zárja be a párbeszédpanelt Bezárás gomb
- most megnézheti a fájl tartalmát a Galaxy-eye (szem) ikonra kattintva. Látni fogja, hogy ez a fájl sok információt tartalmaz az egyes SRA-csatlakozásokról. Ebben a tanulmányban minden csatlakozás egy olyan betegnek felel meg, akinek a mintáit szekvenálták.
a Galaxy képes feldolgozni az összes 2000+ adatkészletet, de ahhoz, hogy ez a bemutató elviselhető legyen, ki kell választanunk egy kisebb részhalmazt. Különösen az ezen adatokkal kapcsolatos korábbi tapasztalataink két érdekes adatkészletet mutatnak SRR11954102és SRR12733957. Szóval, húzzuk ki őket.
megjegyzés Óvakodj a vágásoktól
az alábbi gyakorlati rész a vágóeszközt használja. Két vágott eszköz van a galaxisban történelmi okok miatt. Ez a példa az eszközt használja a teljes névvel vágott oszlopok egy táblázatból (kivágás). Ugyanez a logika vonatkozik a másik eszközre is. Egyszerűen egy kicsit más felülettel rendelkezik.
hands_on Hands-on: adatok részhalmazának létrehozása
- eszköz keresése “kifejezésnek megfelelő sorok kiválasztása” eszköz az eszköz panel szűrő és rendezés szakaszában.
tip tipp: Eszközök keresése
a Galaxy-ban elsöprő mennyiségű eszköz lehet telepítve. Egy adott eszköz megkereséséhez írja be az eszköz nevét az eszközpanel keresőmezőjébe az eszköz megkereséséhez.
- győződjön meg arról, hogy a
SraRunInfo.csvaz imént feltöltött adatkészlet szerepel az eszköz űrlap param-file “sorok kiválasztása” mezőjében.- a “minta” mezőbe írja be a következő kifejezést:
SRR12733957|SRR11954102. Ez két csatlakozás, amelyet a|cső szimbólummal elválasztva szeretnénk megtalálni. A|jelentéseor: aSRR12733957vagySRR11954102tartalmazó sorok keresése.- kattintson a
Executegombra.- Ez létrehoz egy fájlt, amely két sort tartalmaz (nos … az egyik sort fejlécként is használják, így megjelenik a fájl három sorból áll. Semmi baj.)
- vágja ki az első oszlopot a fájlból az eszköz segítségével “kivágás” eszköz, amelyet az eszközablak Szövegmanipulációs szakaszában talál.
- győződjön meg arról, hogy az előző lépésben előállított adatkészlet ki van választva az eszköz űrlap “kivágandó Fájl” mezőjében.
- változtassa meg a “által határolt” értéket
Comma- a “mezők listájában” válassza a
Column: 1lehetőséget.- Hit
Executeez egy szöveges fájlt hoz létre, amely csak két sorból áll:SRR12733957SRR11954102
most, hogy rendelkezünk az adathalmazok azonosítóival, meg kell határoznunk, hogy a le kell töltenünk a tényleges szekvenálási adatokat.
töltse le a szekvenálási adatokat a gyorsabb letöltéssel és a kivonat beolvasásával a FASTQ-ban
hands_on Hands-on: feladat leírása
- gyorsabb letöltés és kivonat beolvasás a FASTQ eszközben a következő paraméterekkel:
- “bemeneti típus kiválasztása”:
List of SRA accession, one per line
- a paraméter param-fájl” sra csatlakozási lista “kell mutatni a kimenet a szerszám” Cut ” az előző lépés.
- kattintson a
Executegombra. Ez futtatja az eszközt, amely beolvassa aSRAadatkészletben felsorolt futások szekvenciaolvasott adatkészleteit. Eltarthat egy ideig. Tehát ez lehet egy jó ideje, hogy nem kap kávét.- számos bejegyzés jön létre az Előzmények panelen, amikor elküldi ezt a munkát:
Pair-end data (fasterq-dump): Párosított végű adatkészleteket tartalmaz (ha vannak ilyenek)Single-end data (fasterq-dump)egyvégű adatkészleteket tartalmaz (ha vannak ilyenek)Other data (fasterq-dump)párosítatlan adatkészleteket tartalmaz (ha vannak ilyenek)fasterq-dump logaz eszköz végrehajtása
az első három elem valójában adatkészletek gyűjteménye. A galaxisban található gyűjtemények az adatkészletek logikai csoportjai,amelyek tükrözik a szemantikai kapcsolatokat a kísérlet / elemzés során. Ebben az esetben az eszköz külön gyűjteményt hoz létre a párosított végű olvasásokhoz, az egyes olvasásokhoz és másokhoz.Lásd a gyűjtemények útmutatók több.
fedezze fel a gyűjteményeket úgy, hogy először a gyűjtemény nevére kattint az Előzmények panelen. Ez bevisz a gyűjteménybe, és megmutatja a benne lévő adathalmazokat. Ezután navigálhat vissza a történelem külső szintjére.
miután fasterq befejezi az adatátvitelt (minden doboz zöld / kész), készen állunk az elemzésre.
most mi van?
mostantól elemezheti a lekért adatokat a Galaxy bármely szekvenciaelemző eszközével és munkafolyamatával. Az SRA minden elképzelhető típusú *-seq kísérlethez biztonsági adatokat tárol.
Ha futtatta ezt az oktatóanyagot, de lekért adatkészleteket, amelyek érdekelték, akkor nézze meg a GTN könyvtár többi részét, hogy ötleteket találjon a Galaxy elemzésére.
Ha azonban letöltötte az oktatóanyag fenti példáiban használt adatkészleteket, akkor készen áll az alábbi SARS-CoV-2 variáns elemzés futtatására.
a SARS-Cov-2 szekvenálási adatok variációs elemzése
az oktatóanyag ezen részében variánshívást és a fent letöltött adatkészletek alapvető elemzését hajtjuk végre. Először letöltjük a Wuhan-Hu-1 SARS-CoV-2 referencia szekvenciát, majd lefuttatjuk az adapter trimmelését, igazítását és variánshívását, és végül megnézzük a talált változatok földrajzi eloszlását.
megjegyzés a usegalaxy.* COVID-19 elemzési projekt
Ez az oktatóanyag az adatok egy részhalmazát használja, és a covid19 variációs elemzési szakaszán fut keresztül.galaxyproject.org.Az adatok covid19.galaxyproject.org folyamatosan frissül, mivel az új adatkészletek nyilvánosságra kerülnek.
Szerezd meg a referencia genomadatokat
a mai referencia genomadatok a SARS-CoV-2, “súlyos akut légzőszervi szindróma koronavírus 2 izolátum Wuhan-Hu-1, teljes genom”, amelynek csatlakozási azonosítója NC_045512.2.
Ezek az adatok a Zenodo-tól érhetők el az alábbi link segítségével.
hands_on Hands-on: Szerezd meg a referencia Genom adatait
importálja a következő fájlt az előzményekbe:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gztipp: Importálás linkeken keresztül
- másolja a link helyét
- nyissa meg a Galaxy Upload Manager (galaxy-Feltöltés az eszköz panel jobb felső sarkában)
- válassza a Beillesztés/Fetch lehetőséget adatok
- illessze be a linket a szövegmezőbe
- nyomja meg a Start gombot
- zárja be az ablakotalapértelmezés szerint a galaxy az URL-t használja névként, ezért nevezze át a fájlokat hasznosabb névvel.
Adaptervágás fastp-vel
a szekvenáló adapterek eltávolítása javítja az illesztéseket és a változathívást. a fastp eszköz automatikusan felismeri a széles körben használt szekvenáló adaptereket.
hands_on Hands-on: feladat leírása
- fastp eszköz a következő paraméterekkel:
- “egyvégű vagy párosított olvasás”:
Paired Collection
- param-file “párosított gyűjtemény (ek) kiválasztása”:
list_paired(a gyorsabb letöltés és kivonat kimenete a FASTQ eszközben olvasható)- a “kimeneti opciók” részben:
- “kimeneti JSON jelentés”:
Yes
igazítás a térképhez a BWA-mem segítségével
a BWA-mem eszköz egy széles körben használt szekvencia-Aligner a rövid olvasású szekvenálási adatkészletekhez, például azokhoz, amelyeket ebben az oktatóanyagban elemezünk.
hands_on Hands-on: Igazítsa a szekvenálást a referencia genomhoz
- térkép a BWA-MEM eszközzel a következő paraméterekkel:
- “kiválaszt egy referencia genomot az előzményekből, vagy beépített indexet használ?”:
Use a genome from history and build index
- param-file “használja a következő adatkészletet referenciaszekvenciaként”:
output(bemeneti adatkészlet)- “egy vagy párosított végolvasás”:
Paired Collection
- param-file “válasszon ki egy párosított gyűjteményt”:
output_paired_coll(a fastp eszköz kimenete)- “állítsa be az olvasási csoportok adatait?”:
Do not set- “válassza ki az elemzési módot”:
1.Simple Illumina mode
távolítsa el a másolatokat a MarkDuplicates segítségével
a MarkDuplicates eszköz eltávolítja a könyvtári előkészítő leletek és szekvenáló leletek. Fontos eltávolítani ezeket a műtárgyi szekvenciákat az egyetlen molekula mesterséges túlreprezentációjának elkerülése érdekében.
hands_on Hands-on: távolítsa el a PCR másolatokat
- MarkDuplicates eszköz a következő paraméterekkel:
- param-file “válassza ki a SAM/BAM adatkészletet vagy adatkészlet-gyűjteményt”:
bam_output(A térkép kimenete a BWA-MEM eszközzel)- “ha igaz, Ne írjon másolatokat a kimeneti fájlba, ahelyett, hogy megfelelő zászlókkal írná őket”:
Yes
igazítási statisztikák létrehozása a samtools statisztikákkal
a fenti duplikált jelölési lépés után statisztikákat generálhatunk az általunk létrehozott IGAZÍTÁSRÓL.
hands_on Hands-on: Igazítási statisztikák létrehozása
- Samtools stats eszköz a következő paraméterekkel:
- param-file “BAM file”:
outFile(a markduplicates eszköz kimenete)- “lefedettségi Eloszlás beállítása”:
No- “kimenet”:
One single summary file- “szűrés Sam zászlók szerint”:
Do not filter- “referencia sorrend használata”:
No- “szűrés régiók szerint”:
No
A lofreq viterbi-vel történő olvasás átrendezése
az újratervezés eszköz kijavítja a Beszúrások és törlések körüli eltéréseket. Ez szükséges a változatok pontos észleléséhez.
hands_on Hands-on: átrendez az indels körül
- átrendez a lofreq eszközzel a következő paraméterekkel:
- param-file “átrendez”:
outFile(a MarkDuplicates eszköz kimenete)- “válassza ki a referencia Genom forrását”:
History
- param-file “Reference”:
output(bemeneti adatkészlet)- az” Advanced options “részben:
- ” hogyan kell kezelni a 2 alapminőségét?”:
Keep unchanged
adjon hozzá indel tulajdonságokat a lofreq insert indel tulajdonságokkal
Ez a lépés hozzáadja az Indel tulajdonságokat az igazítási fájlunkhoz. Erre azért van szükség, hogy a lofreq eszközzel hívási változatokat hívhassunk
hands_on Hands-on: Add indel adottságok
- Insert indel adottságok lofreq eszköz a következő paraméterekkel:
- param-file “olvasás”:
realigned(output of Realign reads tool)- “Indel számítási megközelítés”:
Dindel
- “válassza ki a referencia Genom forrását”:
History
- param-file “referencia”:
output(bemeneti adatkészlet)
hívási változatok a lofreq hívásváltozatok használata
most már készen állunk a változatok hívására.
hands_on Hands-on: Hívásváltozatok
- Hívásváltozatok a lofreq eszközzel a következő paraméterekkel:
- param-file “a bemenet Bam formátumban olvasható”:
output(az Insert indel quality tool kimenete)- “válassza ki a referencia genom”:
History
- param-file “referencia”:
output(bemeneti adatkészlet)- “hívás változatok között”:
Whole reference- “a hívandó változatok típusai”:
SNVs and indels- “Variant hívó paraméterek”:
Configure settings
- a “lefedettség”:
- “minimális lefedettség”:
50- a “Alaphívás”:
- “minimum baseq”:
30- “minimum baseq alternatív bázisokhoz”:
30- a “mapping qualityy
20- “variáns szűrőparaméterek”:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
ennek a lépésnek a kimenete a genom böngészőben megjeleníthető VCF fájlok gyűjteménye.
jegyezze fel a variáns effektusokat az SnpEff eff segítségével:
most kommentáljuk az előző lépésben hívott változatokat a SARS-CoV-2 genomra gyakorolt hatásukkal.
hands_on Hands-on: jegyzetekkel variáns hatások
- SnpEff eff: eszköz a következő paraméterekkel:
- param-file ” szekvencia változások (SNPs, MNPs, InDels)”:
variants(hívásváltozatok kimenete eszköz)- “kimeneti formátum”:
VCF (only if input is VCF)- “CSV jelentés létrehozása, hasznos a downstream elemzéshez (-csvStats)”:
Yes- “annotációs opciók”: `
- “filter output”: `
- “filter out specific effects”:
No
a lépés kimenete egy VCF fájl hozzáadott variáns effektusokkal.
változótáblázat létrehozása SnpSift Kivonatmezők használatával
most különféle effektusokat választunk ki a VCF-ből, és létrehozunk egy táblázatos fájlt, amely könnyebben érthető az emberek számára.
hands_on Hands-on: a változatok táblázatának létrehozása
- SnpSift kivonat mezők eszköz a következő paraméterekkel:
- param-file “variáns bemeneti fájl VCF formátumban”:
snpeff_output(az SnpEff eff: eszköz kimenete)- “kibontandó mezők”:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- “több mező elválasztó”:
,- “üres mezőszöveg”:
.
megnézhetjük a kimeneti fájlokat, és megnézhetjük, hogy a fájlban lévő változatok is le vannak-e írva egy megfigyelhető jegyzetfüzetben, amely megmutatja a földrajzi a SARS-cov-2 variáns szekvenciák eloszlása
érdekes változatok közé tartozik a C-t változat az 14408 pozícióban (14408c/t) az srr11772204-ben, a 28144t/C az srr11597145-ben és a 25563g/t az srr11667145-ben.
összegezze az adatokat a MultiQC-vel
most összefoglaljuk elemzésünket a MultiQC-vel, amely gyönyörű jelentést generál adatainkhoz.
hands_on Hands-on: az adatok összefoglalása
- MultiQC eszköz a következő paraméterekkel:
- Az” eredmények “részben:
- param-repeat” eredmények beszúrása “
- ” melyik eszközt használták naplók létrehozásához?”:
fastp
- param-fájl “a fastp kimenete”:
report_json(a fastp eszköz kimenete)param-repeat “eredmények beszúrása”
- “melyik eszközt használták naplók létrehozása?”:
Samtools
- a “Samtools kimenet”:
- param-repeat “helyezze be a Samtools kimenetet”
- ” a Samtools kimenet típusa?”:
stats
- param-file “Samtools statisztika kimenet”:
output(a Samtools stats eszköz kimenete)param-repeat “eredmények beszúrása”
- “melyik eszközt használták naplók létrehozásához?”:
Picard
- a “Picard kimenet”:
- param-repeat “helyezze be a Picard kimenetet”
- ” a Picard kimenet típusa?”:
Markdups- param-fájl “Picard kimenet”:
metrics_file(a MarkDuplicates eszköz kimenete)param-repeat “Insert Results”
- “melyik eszközt használták naplók létrehozásához?”:
SnpEff
- param-file “kimenet SnpEff”:
csvFile(kimenet SnpEff eff: eszköz)
következtetés
Gratulálunk, most már tudja, hogyan kell importálni a Szekvenciaadatokat az sra-ból, és hogyan kell futtatni egy példaelemzést ezeken az adatkészleteken.
kulcspontok kulcsfontosságú pontok
az SRA Szekvenciaadatai közvetlenül importálhatók a galaxisba
Gyakran Ismételt Kérdések
kérdése van ezzel a bemutatóval kapcsolatban? Nézze meg a VARIÁNSELEMZÉS témakörének GYIK oldalát, hogy megnézze, szerepel-e kérdése ott. Ha nem, kérjük, tegye fel kérdését a GTN Gitter Csatornán vagy a Galaxy Súgófórumon
hasznos irodalom
további információk, beleértve a dokumentációra és az eredeti kiadványokra mutató linkeket, az ebben az oktatóanyagban leírt eszközökkel, elemzési technikákkal és az eredmények értelmezésével kapcsolatban itt találhatók.
visszajelzés
oktatóként használtad ezt az anyagot? Nyugodtan adjon visszajelzést arról, hogyan ment.
idézve ezt az oktatóanyagot
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 az NCBI Sequence Read Archive (sra)-től a Galaxy-ig: SARS-cov-2 variáns elemzés (Galaxy képzési anyagok). / képzés-anyag / témák / változat-elemzés / oktatóanyagok / sars-cov-2 / bemutató.html Online; ma elérhető
- Batut et al., 2018 közösségi alapú adatelemzési képzés a biológiai sejtrendszerek számára 10.1016 / j.cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}