az aminosavakat, nukleotidokat vagy bármely más evolúciós karaktert bizonyos sebességgel helyettesítik. Ha a helyettesítési modell időben visszafordítható, akkor három átmeneti sebesség lesz, a<>B, B<>C és a<>C.
tegyük fel, hogy az arányok 1, 1, illetve 0, helyettesítési egységenként 100 karakter / időegység. Egy időegység után, egy 300 karakteres hosszú szekvenciában, amely eredetileg AS, Bs és Cs-ből állt, arra számítunk, hogy lesz egy A-B helyettesítés és egy B-C helyettesítés. Ha élő organizmusokban két homológ szekvenciát hasonlítunk össze, mivel mindkét szekvenciára egy időegység telt el, akkor két A-tól B-ig, két B-től Csubstitúcióra számíthatunk a mai szekvenciák között.
nem számít, mennyi ideig futtatjuk ezt a folyamatot, soha nem lesz közvetlen helyettesítés a által C. soha nem lesz A-tól C-ig történő helyettesítés az úgynevezett végtelen helyek modell alatt, ahol egynél több helyettesítés nem fordulhat elő egyetlen helyszínen.
mivel azonban a-tól B-ig és B-től C-ig szubsztitúciók gyakoriak, egy véges sitesmodell alatt végül B helyébe C lép egy olyan helyen, ahol a-t korábban B váltotta fel.
a fenti forgatókönyv alapján szimuláltam a szekvencia evolúcióját, 10 egységnyi idő alatt futtatva a szimulációt. Ebből a helyettesítésből megfigyeltem az egyes helyminták következő számait:
| A | B | C | |
|---|---|---|---|
| A | 91 | 9 | 0 |
| B | 5 | 86 | 9 |
| C | 0 | 9 | 91 |
Ezen a viszonylag rövid időtartamon belül nem tűnik úgy, mintha a<>csubstitutions történt volna. Amikor azonban 100 unitsof időre újratervezem a szimulációt:
| A | B | C | |
|---|---|---|---|
| A | 55 | 35 | 10 |
| B | 29 | 36 | 35 |
| C | 20 | 36 | 44 |
mint látható, sok “a” karaktert “C” – re és viceversa-ra cseréltek. Általánosabban, egy véges helyek alatt több szubsztitúciót modelleznek, mert a helyminták eloszlása sokkal laposabbá válik, túl egyszerűen növelve az átlós arányok arányát az átlós számokhoz képest.A PAM és a BLOSUM score mátrixok több szubsztitúciót is figyelembe vesznekradikálisan különböző módon.
az aminosavak PAM mátrixait, valamint a genetikailag kódolt aminosavakhoz használt egyetlen betű rövidítéseket MargaretDayhoff fejlesztette ki. Eredetileg 1978-ban jelentek meg, és a proteinszekvenciák alapján dayhoff az 1960-as évek óta összeállította őket, a Theatlas of Protein Sequence and Structure néven.
A PAM név a “point accepted mutation” – ből származik, és arra utal, hogy egyetlen aminosavat egy másik aminosavval helyettesítenek egy fehérjében.Ezeket a mutációkat úgy azonosítottuk, hogy összehasonlítottuk a nagyon hasonló szekvenciákat legalább 85% – os azonossággal, és feltételezzük, hogy a megfigyelt szubsztitúciók egyetlen mutáció eredménye voltak az ősi szekvencia és az egyik jelenlegi szekvencia között.
A PAM egy időegységet is meghatároz, ahol 1 PAM az az idő, amelyben 1/100 aminosav várhatóan mutáción megy keresztül. A PAM1 valószínűségi mátrix azt mutatja, hogy a J oszlopban lévő aminosav helyébe az I. dayhoff PAM-számaiból számították ki, átméretezve tobe-t 1 PAM időegység. Mint láthatja, a pam1 mátrixban az átlós valószínűségek mind nagyon kicsiek (az összes elemet 10 000-rel skálázták a hitelesség érdekében):
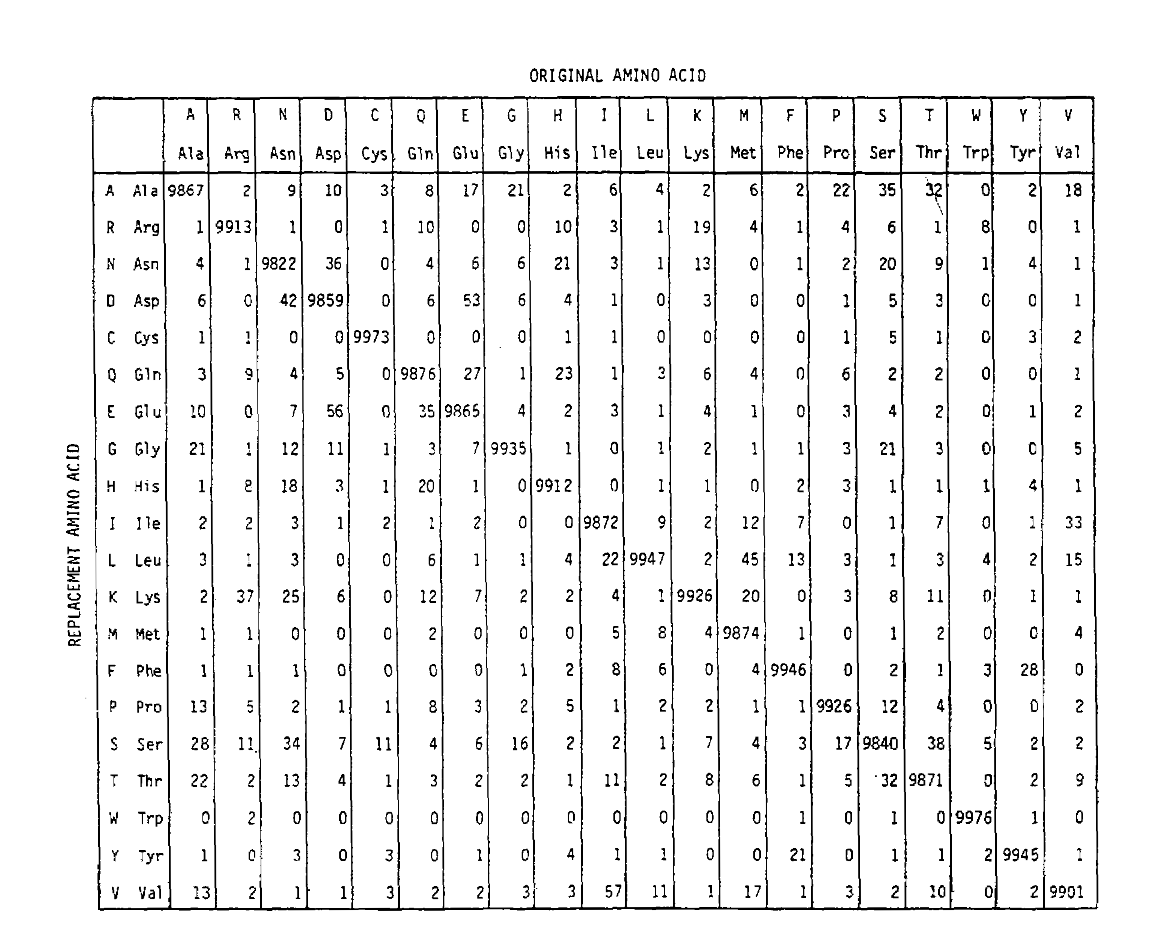
az aminosav-helyettesítési valószínűségek hosszabb időtartamú kiszámításához a mátrix megszorozható önmagával a megfelelő számú alkalommal. Így a PAM250 valószínűségi mátrix, amely leírja a 250 Pam időegységre adott elmozdulási valószínűségeket, a PAM1 valószínűségi mátrix 250 teljesítményre történő emelésével származtatott (az összes elemet 100-mal skáláztákaz olvashatóság érdekében):
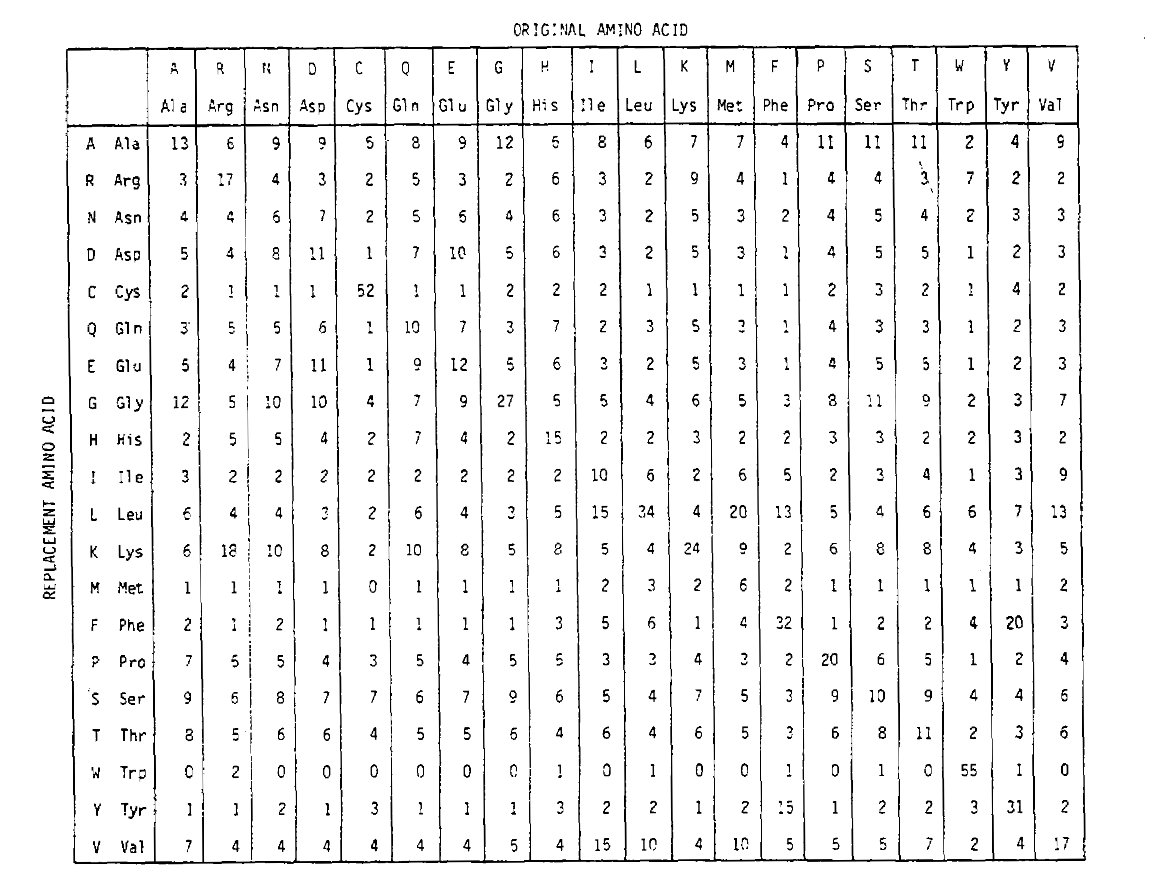
Az ezzel az exponenciációval származtatott helyettesítési valószínűségek több helyettesítést jelentenek. Nem csak az off-diagonalprobabilities arányosan nagyobb, mint amit elvár egy hosszabb timeduration, de ezek laposabb. Például a valin (V) – izoleucin (I) helyettesítés valószínűsége 33-mal nagyobb, mint a V-hisztadin (H)helyettesítés a PAM1 mátrixban, de csak 4,5-rel nagyobb a PAM250 mátrixban.
pontszám mátrixok ezután kiszámítható a valószínűségi mátrixok ésmegfigyelt alapfrekvenciák.
A Steven és Jorja Henikoff által kifejlesztett és 1992-ben megjelent BLOSUM mátrixok egészen más megközelítést alkalmaznak. Míg a PAM implicit módon alkalmazza az evolúció helyhez kötött véges helyek modelljét mátrix hatványozással, a többszörös szubsztitúciók hatását implicit módon kezelik a BLOSUMBAN azáltal, hogy különböző pontszámmátrixokat alakítanak ki különböző időskálákhoz.
a homológ szekvenciák több szekvenciaillesztésén belül konzervált egymással összefüggő aminosavblokkokat azonosítunk. Az egyes blokkokon belül a multiplesequences akkor csoportosul, ha páros átlagos szekvenciaazonosságuk magasabb, mint valamilyen küszöb. A küszöbérték 80% a BLOSUM80 mátrix esetében, 62% A BLOSUM62 esetében, 50% a BLOSUM50 esetében stb.
Ez azt jelenti, hogy a BLOSUM80 esetében a blokkok átlagos páronkénti identitása nem haladja meg a 80% – ot, a BLOSUM62 esetében nem nagyobb, mint 62%, stb.
a homológ szekvenciák aminosav-helyettesítési valószínűségeit a klaszterek közötti páros összehasonlításokból számítjuk ki. Ezek a valószínűségek egyszeri és többszörös szubsztitúciók eredményei lesznek, több szubsztitúcióval, amelyek nagyobb evolúciós távolságokon nagyobb befolyással bírnak. Ezért scorematrices generált páros összehasonlítások klaszterek között átlagosan nagyobb távolság, mint a BLOSUM50 mátrix, természetesen figyelembe thelarger hatása több helyettesítések.
bár különböző útvonalakat választanak, a végső BLOSUM és PAM score matricák valójában nagyon hasonlóak. Henikoff és Henikoff szerint a következő Pam és BLOSUM mátrixok összehasonlíthatók:
| PAM | BLOSUM |
|---|---|
| PAM250 | BLOSUM45 |
| PAM160 | BLOSUM62 |
| PAM120 | BLOSUM80 |
For more information on PAM (Dayhoff) and BLOSUM matrices, see fejezet 2 ofbiológiai szekvencia elemzés Durbin et al. és a Wikipédia.
Update 13 October 2019: a helyettesítési mátrixok másik perspektívájához olvassa el a Compeau és Pevzner bioinformatikai algoritmusok 5.fejezetének (2. vagy 3. kiadás) végén található “kitérők” részt.