målet med denne opplæringen er å introdusere deg til behandlingen av neste generasjons sekvenseringsdata i Galaksen. Denne opplæringen bruker EN COVID – 19 variant ringer Fra Illumina data, men det handler ikke om variant ringer per se.
ved gjennomføringen av denne opplæringen vil du vite:
- hvordan finne data I SRA og overføre denne informasjonen Til Galaxy
- hvordan utføre grunnleggende ngs databehandling I Galaxy inkludert:
- Kvalitetskontroll (QC) Av Illumina data
- Kartlegging
- Fjerning av duplikater
- Variant ringer med
lofreq - Variant merknad
- Bruke datasett samlinger
- Importere data til Jupyter
### Agenda>> i Denne Opplæringen vil vi dekke:>> 1. TOC > {: toc} > {: .agenda} # # to baner gjennom denne opplæringen vi opprettet to baner som du kan følge gjennom denne opplæringen.1. ** Bane 1 * * – start MED NCBIS SRA og søk etter tilgjengelige tiltredelser → Start (#the-sequence-read-archive)2. ** Trajectory 2 * * – bypass NCBI SRA og starte med Galaxy direkte. → Start (#back-in-galaxy) vi anbefaler at Du begynner med **Trajectory 2**.# Sekvensen Lese Arkivethe (https://www.ncbi.nlm.nih.gov/sra ) er den primære arkiv av * umontert leser * for (https://www.ncbi.nlm.nih.gov/). SRA er et flott sted å få sekvenseringsdataene som ligger til grunn for publikasjoner og studier.Denne opplæringen dekker hvordan du får sekvensdata fra SRA til Galakse ved hjelp av en direkte forbindelse mellom de to.> # # # kommentar >> DU vil også høre SRA referert til som * Kort Lese Arkiv*, det opprinnelige navnet.> {:.comment} # # Tilgang SRASRA kan nås enten direkte gjennom sin nettside, eller gjennom verktøypanelet På Galaxy.> ### kommentar>> i Utgangspunktet finnes verktøypanelalternativet for tilgang TIL SRA bare på (https://usegalaxy.org/). Støtte for direkte tilkobling TIL SRA vil bli inkludert i 20.05-utgivelsen Av Galaxy {:.kommentar}> ### hands_on Hands-on: Utforsk SRA Entrez>> 1. Gå Til Din Galaxy-forekomst av valg, for eksempel en av (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) eller noe annet. (Denne opplæringen bruker usegalaxy.org).> 1. Hvis historikken din ikke allerede er tom, kan du starte en ny historikk (se (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) for mer Om Galaksehistorier)> 1. ** Klikk* * `Hent Data ‘ øverst i verktøypanelet.> 1. ** Klikk * * ‘SRA Server’ i listen over verktøy som vises under ‘Hent Data’.> dette tar deg (https://www.ncbi.nlm.nih.gov/sra) – du kan også starte direkte fra SRA. En søkeboks vises øverst på siden. Prøv å søke etter noe du er interessert i, for eksempel `dolphin` eller `kidney` eller `dolphin kidney` og deretter * * klikk * * På ‘Search’ – knappen.>> dette returnerer en liste over * Sra-Eksperimenter * som samsvarer med søkestrengen din. Sra-Eksperimenter, også kjent som *SRX-oppføringer*, inneholder sekvensdata fra et bestemt eksperiment, samt en forklaring på selve eksperimentet og andre relaterte data. Du kan utforske de returnerte forsøkene ved å klikke på navnet deres. Se (https://www.ncbi.nlm.nih.gov/bøker / NBK56913/) i (https://www.ncbi.nlm.nih.gov/ bøker/n/ helpsrakb/) for mer.>>når du skriver inn tekst i SRA-søkeboksen, bruker du (https://www.ncbi.nlm.nih.gov/sra /docs/srasearch/). Entrez støtter både enkle tekstsøk og svært presise søk som sjekker bestemte metadata og bruker vilkårlig komplekse logiske uttrykk. Entrez lar deg skalere opp søkene fra grunnleggende til avansert som du begrense søkene. Syntaksen til avanserte søk kan virke skremmende, MEN SRA gir en grafisk (https://www.ncbi.nlm.nih.gov/sra/advanced/) for å generere den spesifikke syntaksen. OG som vi skal se nedenfor, GIR Sra Run Selector et enda vennligere brukergrensesnitt for å begrense våre valgte data.>> Lek deg rundt MED SRA Entrez-grensesnittet, inkludert advanced query builder, for å se om du kan identifisere ET sett MED sra-eksperimenter som er relevante for et av dine forskningsområder.{: .hands_on} > ### hands_on Hands-on: Generer liste over matchende eksperimenter ved Hjelp Av Entrez>> nå som DU har en grunnleggende kjennskap TIL SRA Entrez, la oss finne sekvensene som brukes i denne opplæringen.>> 1. Hvis du ikke allerede er der, **naviger * * tilbake til (https://www.ncbi.nlm.nih.gov/sra> 1. ** Clear * * enhver søk tekst fra søkeboksen.> 1. ** Skriv * * ‘sars-cov-2′ i søkeboksen og * * klikk * * `Søk’.> dette returnerer en lang liste OVER sra-eksperimenter som samsvarer med søket vårt, og den listen er altfor lang til å bruke i en opplæringsøvelse. På dette punktet kunne vi bruke advanced Entrez query builder vi lærte om ovenfor.> Men Vi vil Ikke. I Stedet kan sende *for lenge for en tutorial * liste resultatene vi har TIL Sra Run Velgeren, og bruke sin vennligere grensesnitt for å begrense våre resultater.>> !(../../bilder / sra_entrez.png){: .hands_on} > ### hands_on Hands-on: Gå fra Entrez Til Sra Run Selector>> vis resultater som en utvidet interaktiv tabell ved Hjelp Av RunSelector.>> 1. Klikk Send resultater For Å Kjøre velgeren, som vises i en boks øverst i søkeresultatene.>> !(../../bilder / sra_entrez_result.png)>>> # # # tips hva om Du ikke ser Koblingen Kjør Velger?>>>> du har kanskje lagt merke til denne teksten tidligere da Du utforsket Entrez-søk. Denne teksten vises bare en del av tiden, når antall søkeresultater faller innenfor et ganske bredt vindu. Du vil ikke se det hvis du bare har noen få resultater, og du vil ikke se det hvis du har flere resultater enn Kjør Velgeren kan godta.>>>> Du må Kjøre Velgeren for å sende resultatene dine til Galaksen.* Hva om du ikke har nok resultater til å utløse denne linken blir vist? I så fall kaller du komme Til Kjør Velgeren ved * * klikke * * på` Send til ‘ nedtrekksmenyen øverst til høyre i resultatpanelet. For Å Komme Til Run Selector, * * velg * * ‘Run Selector’ og deretter * * klikk * * På` Go ‘ – knappen.> !(../../bilder / sra_entrez_send_to.> {: .tips} >>> 1. ** Klikk* * `Send resultater For Å Kjøre velgeren ‘ øverst i søkeresultatpanelet. (Hvis du ikke ser denne linken, så se kommentaren rett over.){: .hands_on}# # Sra Run Selectorvi lærte tidligere å begrense søkeresultatene ved å bruke Entrezs avanserte syntaks. Men vi utnyttet ikke den kraften da vi var i Entrez. I stedet brukte vi et enkelt søk og sendte deretter alle resultatene til Run Selector. Vi har ennå ikke den (korte) listen over resultater vi ønsker å kjøre analyse på. * Hva gjør vi?* Bruk Entrez grensesnitt for å begrense resultatene ned til en størrelse Som Kjør Velgeren kan konsumere . * Bruk Run Selector er mye vennligere grensesnitt til 1. Lettere forstå dataene vi har 1. Begrense disse resultatene ved hjelp av denne kunnskapen.> ### kommentar Run Selector er både mer og mindre enn Entrez>> Run Selector kan gjøre det meste, men ikke alt Av Hva Entrez search syntaks kan gjøre. Run selector bruker * fasettert søk * teknologi som er enkel å bruke, og kraftig, men som har iboende grenser. Spesielt Vil Entrez fungere bedre når du søker på attributter som har titalls, hundrevis eller tusenvis av forskjellige verdier. Kjør Velgeren vil fungere bedre søker attributter med færre enn 20 forskjellige verdier. Heldigvis, som beskriver de fleste søk.{: .comment}Kjør Velgeren vinduet er delt inn i flere paneler:** * ` Filters List’**: i øvre venstre hjørne. Det er her vi vil avgrense søket vårt.* * * ‘Select’**: et sammendrag av hva som først ble sendt Til Run Selector, og hvor mye av det vi har valgt så langt. (Og så langt har vi ikke valgt noe av det.) Legg også merke til den tantalizing `men fortsatt gråtonet, ‘Galaxy’ – knappen.** * ` Found x Items ‘ * * I Utgangspunktet er dette listen over elementer sendt Til Run Selector Fra Entrez. Denne listen vil krympe når vi bruker filtre til den.!(../../bilder / sra_run_selector.png)> ### kommentar hvorfor gikk antall funnet elementer *opp?* >> Husk at Entrez-grensesnittet viser sra-eksperimenter (SRX-oppføringer). Run Selector lister * runs * – sekvensering datasett – og det er * en eller flere * kjører per eksperiment. Vi har de samme dataene som før, vi ser nå bare det i finere detalj.{: .Filterlisten øverst til venstre viser kolonner i våre resultater som enten har kontinuerlige tallverdier, eller 10 eller mindre (du kan endre dette tallet) distinkte verdier i dem. ** Bla * * ned gjennom listen velg noen av filtrene. Når et filter er valgt, vises en* verdier * – boks nedenfor, oppføringsalternativer for dette filteret og antall kjøringer med hvert alternativ. Disse verdiene / alternativene trekkes fra datasettmetadataene. Prøv * * velge * * noen interessante klingende filtre og deretter * * velg * * ett eller flere alternativer for hvert filter. Prøv * * opphev valget av * * alternativer og filtre. Når du gjør dette, vil antall funnet resultater reduseres eller øke.> ### tips Tips: Bruk Filtre for å bedre forstå dataene>>Filtre er hvordan du begrenser datasettene som vurderes for å sende Til Galaxy, men De er også en utmerket måte å forstå dataene dine på:> først er det å velge et filter en enkel måte å se verdiområdet i en kolonne. Du kan kanskje ikke (https://www.google.com/search?q=sra+sirs_outcome), men du kan muligens finne ut det ved å se hvilke verdier som er i den.> For Det Andre kan du utforske hvordan ulike kolonner forholder seg til hverandre. Er det en sammenheng mellom ‘sirs_outcome’ verdier og ‘disease_stage’ verdier?{: .tips} > # # # hands_on Hands-on: Begrense resultatene ved Hjelp Av Kjør Velgeren>> 1. Hvis du har noen filtre slått på,** fjern markeringen * * dem.> når du har gjort dette, vil det ikke være noen* verdier * bokser som vises under Filterlisten.> 2. ** Kopier og lim inn * * denne søkestrengen i søkeboksen` Funnet Elementer’.>> SRR11772204 ELLER SRR11597145 eller SRR11667145>> denne hånden-plukket sett med løyper begrenser våre resultater til 3 løyper fra ulike geografiske distribusjon.{: .hands_on}dette reduserer Listen ‘Funnet Elementer’ fra titusenvis av løp til 3 løp (et håndterbart nummer for en opplæring!). Men Vi er ikke helt ferdig med Run Selector ennå. Merk at `Galaxy ‘ – knappen fortsatt er nedtonet. Vi har innsnevret våre alternativer, men vi har faktisk ikke valgt noe å sende Til Galaxy ennå.Det er mulig å velge hver gjenværende kjøre av * * klikke * * haken øverst i den første kolonnen. Du kan velge bort alt ved å * * klikke * * på `X’.> ### hands_on Hands-on: Velg kjøringer og send Til Galakse>> 1. Velg alle kjører ved * * klikke * * den`X’.> Og Nå er «Galaxy» – knappen live.> 1. ** Klikk * * På ‘Galaxy’ – knappen i ‘Select’ – delen øverst på siden.{: .hands_on}# # Tilbake I GalaxyWhen vi klikker ‘Galaxy’ I Run Selector flere ting skje. Først lanserer det en ny nettleserfane eller et vindu som åpnes I Galaxy. Du vil se * big green box * som indikerer at håndtrykket mellom Sra og Galaxy var vellykket, og du vil da se en ny ‘ SRA ‘ jobb i historikkpanelet. Denne boksen kan starte som grå / ventende, noe som indikerer at overføringen ennå ikke har startet, eller det kan gå rett til gul / kjører eller til grønn / ferdig.> ### hands_on Hands-on: Undersøk det nye Sra-Datasettet>> 1. Når sra-overføringen er fullført, * * klikk * * på datasettets galaxy-eye (eye) – ikon.>> dette viser datasettet i Galaksens midtpanel.{: .hands_on} ‘ SRA ‘ datasettet er ikke sekvensdata ,men heller * metadata * som vi vil bruke til å få sekvensdata fra SRA. Denne metadataen speiler informasjonen vi så i Run Selector` S ‘Found Items’ – delen. Metadataene er ikke sluttdataene vi søker FRA SRA, men å ha alle disse metadataene er ofte nyttige i etterfølgende analysestrinn.La oss nå bruke metadataene til å hente sekvensdataene fra SRA. SRA gir verktøy for å trekke ut all slags informasjon, inkludert sekvensdataene selv. Galaxy Tool ‘Raskere Nedlasting Og Utdrag Leser I FASTQ’ er basert på sra (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump) verktøyet, og gjør nettopp det.– >
- Finn nødvendige data i SRA
- hands_on Hands-on: Oppgavebeskrivelse
- kommentar Kommentar
- Prosess og filter SraRunInfo.csv-fil i Galaksen
- hands_on Hands-on: Last Opp SraRunInfo.csv-fil Til Galaxy
- kommentar Pass På Kutt
- hands_on Hands-on: Opprette et delsett av data
- tips Tips: Finne verktøy
- Last ned sekvenseringsdata Med Raskere Nedlasting Og Utdrag Leser I FASTQ
- hands_on Hands-on: Oppgave beskrivelse
- Hva nå?
- Variasjonsanalyse AV sars-Cov-2 sekvenseringsdata
- kommenter brukergalaksen.* COVID-19 analyseprosjekt
- Få referanse genom data
- hands_on Hands-on: Få referanse genomet data
- Tips: Importerer via lenker
- adaptertrimming med fastp
- hands_on Hands-on: Oppgave beskrivelse
- justering med kart med bwa-mem
- hands_on Hands-on: Juster sekvensering leser til referansegenom
- Fjern duplikater med MarkDuplicates
- hands_on Hands-on: Fjern pcr duplikater
- generer justeringsstatistikk med samtools stats
- hands_on Hands-on: Generere justering statistikk
- juster leser verktøyet korrigerer feiljusteringer rundt innsettinger og slettinger. Dette kreves for å kunne oppdage varianter nøyaktig. hands_on Hands-on: Realign leser rundt indels Realign leser med lofreq verktøy med følgende parametere: param-fil «Leser til realign»:outFile (utgang Av MarkDuplicates verktøy) «Velg kilden til referansegenomet»: History param-fil «Referanse»:output (Input datasett) I «Avanserte alternativer»: «hvordan håndtere basekvaliteter av 2?»: Keep unchanged Legg indel kvaliteter med lofreq Sett inn indel kvaliteter
- hands_on Hands-on: Legg indel kvaliteter
- samtalevarianter bruke lofreq samtale varianter
- hands_on Hands-on: Call varianter
- Annotere varianteffekter Med SnpEff eff:
- hands_on Hands-on: Kommentere variant effekter
- Lag tabell med varianter ved Hjelp Av SnpSift Extract Fields
- hands_on Hands-on: Lag tabell med varianter
- Oppsummer data Med MultiQC
- hands_on Hands-on: Oppsummere data
- konklusjon
- nøkkelpunkter
- Ofte Stilte Spørsmål
- Nyttig litteratur
- Tilbakemelding
- Siterer Denne Opplæringen
- details BibTeX
Finn nødvendige data i SRA
Først må vi finne et godt datasett å leke med. Sequence Read Archive (SRA) er det primære arkivet for umonterte lesninger som drives AV US National Institutes Of Health (NIH). SRA er et flott sted å få sekvenseringsdataene som ligger til grunn for publikasjoner og studier. La oss gjøre det:
hands_on Hands-on: Oppgavebeskrivelse
- Gå TIL NCBIS SRA-side ved å peke nettleseren din til https://www.ncbi.nlm.nih.gov/sra
- i søkeboksen skriv inn
SARS-CoV-2 Patient Sequencing From Partners / MGH(Alternativt klikker du bare på denne linken)
- websiden vil vise et stort antall SRA datasett(i skrivende stund var det 2.223). Dette er data fra en studie som beskriver analyse AV SARS-CoV-2 I Boston-omradet.
- Last ned metadata som beskriver disse datasettene ved å:
- klikke På Send til: dropdown
- Velge
FileEndre Format til
RunInfo- Klikk På Opprett filher er hvordan Det skal se ut:
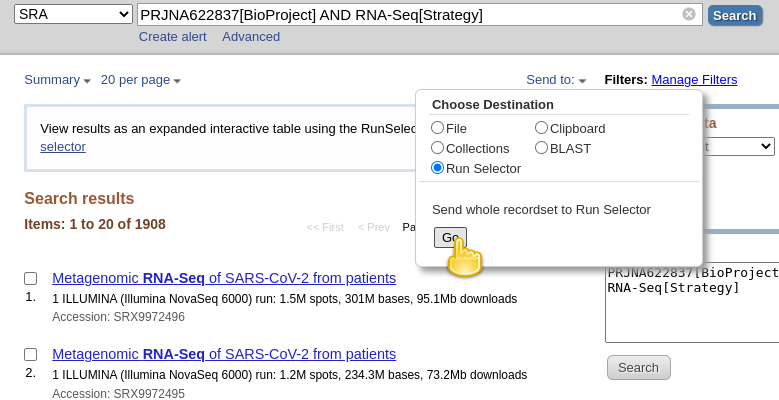
- Dette vil skape en ganske stor
SraRunInfo.csvfil iDownloadsmappe.
Nå som vi har lastet ned denne filen, kan vi gå til En Galakse-forekomst og begynne å behandle den.
kommentar Kommentar
Merk at filen vi nettopp lastet ned ikke sekvenserer data selv. Snarere er det metadata som beskriver egenskapene til sekvensering leser. Vi vil filtrere denne listen ned til bare noen få tiltredelser som vil bli brukt i resten av denne opplæringen.
Prosess og filter SraRunInfo.csv-fil i Galaksen
hands_on Hands-on: Last Opp SraRunInfo.csv-fil Til Galaxy
- Gå Til Din Galaxy-forekomst av valg, for eksempel en Av usegalaxy.org, usegalaxy.eu, usegalaxy.org.au eller noe annet. (Denne opplæringen bruker usegalaxy.org).
- Klikk Last Opp Data-knappen:
- i dialogboksen som vises, klikk på «Velg lokale filer»-knappen:

- Finn Og velg
SraRunInfo.csvfil fra datamaskinen- Klikk Startknappen
- Lukk dialogboksen Ved å trykke på lukk knapp
- du kan nå se på innholdet I denne filen ved å klikke galaxy-eye (eye) ikon. Du vil se at denne filen inneholder mye informasjon om individuelle sra-tiltredelser. I denne studien tilsvarer hver tiltredelse en individuell pasient hvis prøver ble sekvensert.
Galaxy kan behandle alle 2000 datasett, men for å gjøre denne opplæringen utholdelig må vi velge en mindre delmengde. Spesielt vår tidligere erfaring med disse dataene viser to interessante datasett SRR11954102 og SRR12733957. Så, la oss trekke dem ut.
kommentar Pass På Kutt
Den Praktiske delen nedenfor bruker Kuttverktøy. Det er to kuttverktøy I Galaxy på grunn av historiske årsaker. Dette eksemplet bruker verktøy med hele navnet Klipp ut kolonner fra en tabell (klipp ut). Den samme logikken gjelder imidlertid for det andre verktøyet. Den har bare et litt annet grensesnitt.
hands_on Hands-on: Opprette et delsett av data
- Finn verktøy «Velg linjer som samsvarer med et uttrykk» verktøy I Filter og Sorter delen av verktøypanelet.
tips Tips: Finne verktøy
Galaxy kan ha en overveldende mengde verktøy installert. For å finne et bestemt verktøy skriv inn verktøynavnet i verktøypanelets søkeboks for å finne verktøyet.
- Kontroller at
SraRunInfo.csvdatasettet vi nettopp lastet opp, er oppført i param-filen «Velg linjer fra» – feltet i verktøyskjemaet.- i feltet «mønster» skriver du inn følgende uttrykk →
SRR12733957|SRR11954102. Dette er to tiltredelser vi ønsker å finne adskilt av rørsymbolet||betyror: finn linjer som inneholderSRR12733957ellerSRR11954102.- Klikk
Executeknapp.- dette vil generere en fil som inneholder to linjer ( vel … en linje brukes også som topptekst, så det vises at filen har tre linjer. Det er OK.)
- Klipp den første kolonnen fra filen ved hjelp av verktøyet » Cut » verktøy, som du finner I Tekstmanipulering delen av verktøyruten.
- Kontroller at datasettet produsert av forrige trinn er valgt i feltet «Fil å kutte» i verktøyskjemaet.
- Endre «Avgrenset av» til
Comma- i» liste over felt » velg
Column: 1.- Hit
ExecuteDette vil produsere en tekstfil med bare to linjer:SRR12733957SRR11954102
Nå som vi har identifikatorer av datasett vi vil vi trenger å laste ned de faktiske Sekvenseringsdataene.
Last ned sekvenseringsdata Med Raskere Nedlasting Og Utdrag Leser I FASTQ
hands_on Hands-on: Oppgave beskrivelse
- Raskere Nedlasting Og Utdrag Leser I FASTQ verktøy med følgende parametere:
- «velg inngangstype»:
List of SRA accession, one per line
- parameteren param-fil «sra tiltredelsesliste» skal peke utgangen av verktøyet «Cut» fra forrige trinn.
- Klikk på
Execute– knappen. Dette vil kjøre verktøyet, som henter sekvensen lese datasett for kjøringer som ble oppført iSRAdatasett. Det kan ta litt tid. Så dette kan være en god tid å få kaffe.- Flere oppføringer opprettes i historikkpanelet når du sender inn denne jobben:
Pair-end data (fasterq-dump): Inneholder Sammenkoblede datasett (hvis tilgjengelig)
Single-end data (fasterq-dump)Inneholder Enkelt-end datasett (hvis tilgjengelig)
Other data (fasterq-dump)Inneholder Uparede datasett (hvis tilgjengelig)fasterq-dump logInneholder Informasjon om verktøyutførelse
de tre første elementene er faktisk samlinger av datasett. Samlinger I Galaksen er logiske grupperinger av datasett som reflekterer de semantiske relasjonene mellom Dem i eksperimentet / analysen. I dette tilfellet verktøyet oppretter en egen samling hver for paret-end leser, enkelt leser, og andre.Se Samlinger tutorials for mer.
Utforsk samlingene ved først å klikke på samlingsnavnet i historikkpanelet. Dette tar deg inne i samlingen og viser deg datasettene i den. Du kan deretter navigere tilbake til det ytre nivået av historien din.
Når fasterq fullfører overføring av data (alle boksene er grønne / ferdige), er vi klare til å analysere det.
Hva nå?
du kan nå analysere de hentede dataene ved hjelp av sekvensanalyseverktøy og arbeidsflyter I Galaxy. SRA holder backing data for alle tenkelige type * – seq eksperiment.
hvis du kjørte denne opplæringen, men hentet datasett som du var interessert i, så se resten AV GTN biblioteket for ideer om hvordan å analysere I Galaxy.
men hvis du hentet datasettene som ble brukt i denne opplæringens eksempler ovenfor, er DU klar til å kjøre sars-CoV-2 variantanalysen nedenfor.
Variasjonsanalyse AV sars-Cov-2 sekvenseringsdata
i denne delen av opplæringen vil vi utføre variant kall og grunnleggende analyse av datasettene lastet ned ovenfor. Vi starter med å laste Ned Wuhan-Hu-1 sars-CoV-2 referansesekvens, deretter kjøre adapter trimming, justering og variant ringer og til slutt se på den geografiske fordelingen av noen av de funnet varianter.
kommenter brukergalaksen.* COVID-19 analyseprosjekt
denne opplæringen bruker et delsett av dataene og går gjennom variation Analysissection av covid19.galaxyproject.org.Dataene for covid19.galaxyproject.org isbeing oppdateres kontinuerlig som nye datasett blir offentliggjort.
Få referanse genom data
referansen genom data for i dag er FOR SARS-CoV-2, «Alvorlig akutt respiratorisk syndrom coronavirus 2 isolere Wuhan-Hu-1, komplett genom», å ha tiltredelse ID AV NC_045512.2.
Disse dataene er tilgjengelige Fra Zenodo ved å bruke følgende lenke.
hands_on Hands-on: Få referanse genomet data
Importer følgende fil inn i din historie:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzTips: Importerer via lenker
- Kopier lenkeplasseringen
- Åpne Galaxy Upload Manager (galaxy-opplasting øverst til høyre på verktøypanelet)
- Velg Lim Inn/Hent data
- lim inn lenken i tekstfeltet
- trykk på start
- lukk vinduetsom standard bruker galaxy url som navn, så omdøpe filene med et mer nyttig navn.
adaptertrimming med fastp
Fjerning av sekvenseringsadaptere forbedrer justeringer og variantanrop. fastp tool kan automatisk oppdage mye brukt sekvensering adaptere.
hands_on Hands-on: Oppgave beskrivelse
- fastp verktøy med følgende parametere:
- «Single-end eller sammenkoblet leser»:
Paired Collection
- param-fil «Velg sammenkoblede samling (er)»:
list_paired(utdata Av Raskere Nedlasting Og Utdrag Leser I FASTQ-verktøyet)
justering med kart med bwa-mem
bwa-mem tool er en mye brukt sekvens aligner For Kort Lese sekvensering datasett som de VI ANALYSERER i denne opplæringen.
hands_on Hands-on: Juster sekvensering leser til referansegenom
- Kart MED bwa-mem-verktøyet med følgende parametere:
- «Vil du velge et referansegenom fra historien din eller bruke en innebygd indeks?»:
Use a genome from history and build index
- param-fil «Bruk følgende datasett som referansesekvens»:
output(Input datasett)- «enkel eller Sammenkoblet-end leser»:
Paired Collection
- param-fil «velg en sammenkoblet samling»:
output_paired_coll(utdata fra fastp-verktøyet)- «sett lese grupper informasjon?»:
Do not set- «Velg analysemodus»:
1.Simple Illumina mode
Fjern duplikater med MarkDuplicates
MarkDuplicates verktøyet fjerner dupliserte sekvenser som stammer fra bibliotek forberedelse artefakter og sekvensering artefakter. Det er viktig å fjerne disse artefaktuelle sekvensene for å unngå kunstig overrepresentasjon av enkeltmolekyl.
hands_on Hands-on: Fjern pcr duplikater
- MarkDuplicates verktøy med følgende parametere:
- param-file «Select SAM/bam dataset or dataset collection»:
bam_output(utdata Av Kart med bwa-mem-verktøy)- «hvis sann ikke skriv duplikater til utdatafilen i stedet for å skrive dem med passende flaggsett»:
Yes
generer justeringsstatistikk med samtools stats
etter duplikatmerkingstrinnet ovenfor Kan Vi Generere statistikk OM JUSTERINGEN VI har generert.
hands_on Hands-on: Generere justering statistikk
- Samtools statistikk verktøy med følgende parametere:
- param-fil «BAM-fil»:
outFile(utgang Av MarkDuplicates verktøy)- «Set dekning distribusjon»:
No- «Output»:
One single summary file- «filtrer etter sam-flagg»:
Do not filter- «bruk en referansesekvens»:
No- «filtrer etter regioner»:
No
juster leser verktøyet korrigerer feiljusteringer rundt innsettinger og slettinger. Dette kreves for å kunne oppdage varianter nøyaktig.
hands_on Hands-on: Realign leser rundt indels
- Realign leser med lofreq verktøy med følgende parametere:
- param-fil «Leser til realign»:
outFile (utgang Av MarkDuplicates verktøy)
- «Velg kilden til referansegenomet»:
History
- param-fil «Referanse»:
output (Input datasett)
- I «Avanserte alternativer»:
- «hvordan håndtere basekvaliteter av 2?»:
Keep unchanged
Legg indel kvaliteter med lofreq Sett inn indel kvaliteter
hands_on Hands-on: Realign leser rundt indels
- Realign leser med lofreq verktøy med følgende parametere:
- param-fil «Leser til realign»:
outFile(utgang Av MarkDuplicates verktøy) - «Velg kilden til referansegenomet»:
History- param-fil «Referanse»:
output(Input datasett)
- param-fil «Referanse»:
- I «Avanserte alternativer»:
- «hvordan håndtere basekvaliteter av 2?»:
Keep unchanged
- «hvordan håndtere basekvaliteter av 2?»:
- param-fil «Leser til realign»:
dette trinnet legger indel kvaliteter i vår justering fil. Dette er nødvendig for å ringe varianter ved Hjelp Av Anropsvarianter med lofreq tool
hands_on Hands-on: Legg indel kvaliteter
- Sett indel kvaliteter med lofreq verktøy med følgende parametere:
- param-fil «Leser»:
realigned(utgang Av Realign leser verktøy)- «Indel beregning tilnærming»:
Dindel
- «Velg kilden for referansegenomet»:
History
- param-fil «referanse»:
output(input datasett)
samtalevarianter bruke lofreq samtale varianter
vi er nå klar til å ringe varianter.
hands_on Hands-on: Call varianter
- Call varianter med lofreq verktøy med følgende parametere:
- param-fil «Input leser I bam format»:
output(utgang Av Insert indel kvaliteter verktøy)- «Velg kilden for referanse genom»:
History
- param-fil «Referanse»:
output(input datasett)- «kall varianter over»:
Whole reference- «typer varianter å ringe»:
SNVs and indels- «variant ringer parametere»:
Configure settings
- I «Dekning»:
- «Minimal dekning»:
50I «Base-ringer»:
- «minimum baseq»:
30«minimum baseq for alternative baser»:
30- «variantfilterparametere»:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
utgangen av dette trinnet er en samling AV VCF-filer som kan visualiseres i en genomleser.
Annotere varianteffekter Med SnpEff eff:
Vi vil nå annotere varianter vi ringte i forrige trinn med effekten de har på sars-CoV-2 genomet.
hands_on Hands-on: Kommentere variant effekter
- SnpEff eff: verktøy med følgende parametere:
- param-fil » Sekvensendringer (SNPs, MNPs, InDels)»:
variants(utgang Av Anropsvarianter verktøy)- «Utdataformat»:
VCF (only if input is VCF)- «Opprett CSV-rapport, nyttig for nedstrøms analyse (- csvStats)»:
Yes- «Annotasjonsalternativer»: `
- «filter output»: `
- «filtrer ut spesifikke effekter»:
No
utgangen av dette trinnet er en vcf-fil med ekstra varianteffekter.
Lag tabell med varianter ved Hjelp Av SnpSift Extract Fields
Vi vil nå velge ulike effekter FRA VCF og lage en tabellfil som er lettere å forstå for mennesker.
hands_on Hands-on: Lag tabell med varianter
- SnpSift Pakk Felt verktøy med følgende parametere:
- param-fil «Variant input fil I VCF format»:
snpeff_output(utgang Av SnpEff eff: verktøy)- «felt å trekke ut»:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- «flere feltskilletegn»:
,- «tomt felt tekst»:
.
Vi kan inspisere utdatafilene og se om Varianter i denne filen også er beskrevet i en observerbar notatbok som viser den geografiske fordeling av sars-cov-2 variantsekvenser
interessante varianter inkluderer c til t-varianten i posisjon 14408 (14408c/t) i srr11772204, 28144t/c i srr11597145 og 25563g / t i srr11667145.
Oppsummer data Med MultiQC
vi vil nå oppsummere vår analyse Med MultiQC, som genererer en vakker rapport for våre data.
hands_on Hands-on: Oppsummere data
- MultiQC verktøy med følgende parametere:
- I «Resultater»:
- param-repeat » Sett Inn Resultater «
- » hvilket verktøy ble brukt generere logger?»:
fastp
- param-fil «Utdata av fastp»:
report_json(utdata fra fastp-verktøyet)param-repeat» Sett Inn Resultater «
- » hvilket verktøy ble brukt til å generere logger?»:
Samtools
- I «Samtools utgang»:
- param-gjenta «Sett Inn Samtools utgang»
- «Type Samtools utgang?»:
stats
- param-fil «samtools stats output»:
output(utdata Av samtools statistikkverktøy)param-repeat «Sett Inn Resultater»
- «hvilket verktøy ble brukt generere logger?»:
Picard
- I «Picard-utgang»:
- param-gjenta «Sett Inn Picard-utgang»
- «Type Picard-utgang?»:
Markdups- param-fil «Picard-utgang»:
metrics_file(utdata Av MarkDuplicates verktøy)param-repeat «Sett Inn Resultater»
- «hvilket verktøy ble brukt generere logger?»:
SnpEff
- param-fil «Utgang Av SnpEff»:
csvFile
konklusjon
gratulerer, du vet nå hvordan du importerer Sekvensdata fra sra og hvordan du kjører en eksempelanalyse på disse datasettene.
nøkkelpunkter
Sekvensdata I Sra kan importeres direkte til Galaksen
Ofte Stilte Spørsmål
Har du spørsmål om denne opplæringen? Sjekk UT FAQ-siden FOR Variantanalyseemnet for å se om spørsmålet ditt er oppført der. Hvis ikke, kan du stille spørsmålet ditt PÅ GTN Gitter Channel eller Galaxy Help Forum
Nyttig litteratur
Ytterligere informasjon, inkludert lenker til dokumentasjon og originale publikasjoner, om verktøy, analyseteknikker og tolkning av resultatene beskrevet i denne opplæringen finner du her.
Tilbakemelding
brukte du dette materialet som instruktør? Føl deg fri til å gi oss tilbakemelding på hvordan det gikk.
Siterer Denne Opplæringen
- Marius van Den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 FRA NCBIS Sekvens Lese Arkiv (SRA) Til Galaxy: SARS-CoV-2 variant analyse (Galaxy Opplæringsmateriell). / trening-materiale / emner / variant-analyse/oppl ring/sars-cov-2 / tutorial.html Online; tilgjengelig I DAG
- Batut et al., 2018 Fellesskapsdrevet Dataanalyse Trening For Biologi Cellesystemer 10.1016 / j. cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}