Aminosyrer, nukleotider eller annen evolusjonær karakter erstattes av andre med en viss hastighet. Tenk deg for eksempel en evolusjonær sekvens Med tremulige tilstander, A, B og C. hvis substitusjonsmodellen er tids-reversibel, vil det være tre overgangshastigheter, A<>B, B<>c og a<>c.
Anta at satsene er henholdsvis 1, 1 og 0 i substitusjonsenheter per 100tegn per tidsenhet. Etter en tidsenhet, i en 300 tegn longsequence opprinnelig sammensatt like Av As, Bs og Cs, forventer vi at det skal havebeen en a Til b substitusjon og En B Til C substitusjon. Hvis vi sammenligner to homologe sekvenser i levende organismer, fordi en tidsenhet har passert for begge sekvenser, forventer vi to A Til B og to B Til Csubstitusjoner mellom dagens sekvenser.
Uansett hvor lenge vi kjører denne prosessen for, vil det aldri bli en direkteplassering Av A av C. det vil heller aldri være En a Til C substitusjon under aso – kalt uendelig nettsteder modell, hvor ikke mer enn en substitusjon kan forekomme på et enkelt nettsted.Men Siden a Til B og b Til C substitusjoner er vanlig, under en endelig sitesmodel slutt b vil bli erstattet Av C på et sted Hvor A ble tidligere erstattet Av B. denne indirekte erstatning Av A Med C (eller ekvivalent i atime-reversibel modell, C Av A) blir mer sannsynlig jo lengre tidsperiodeseparere homologe sekvenser.
jeg simulerte sekvensutvikling basert på scenariet ovenfor, kjører thesimulering for 10 tidsenheter. Fra denne substitusjon observert jeg thefollowing teller for hvert område mønster:
| A | B | C | |
|---|---|---|---|
| A | 91 | 9 | 0 |
| B | 5 | 86 | 9 |
| C | 0 | 9 | 91 |
innenfor denne relativt korte varigheten ser det ikke ut som om noen en<>csubstitutions har oppstått. Men når jeg reran simuleringen for 100 unitsof tid:
| A | B | C | |
|---|---|---|---|
| A | 55 | 35 | 10 |
| B | 29 | 36 | 35 |
| C | 20 | 36 | 44 |
som du kan se, har mange «a» – tegn blitt erstattet med «c» og omvendt. Mer generelt, under en endelig nettsteder modell flere substitutionscause fordelingen av nettstedet mønster teller til å bli mye flatere beyondsimply øke andelen av off-diagonal i forhold til diagonale teller.PAM og BLOSUM score matriser står for flere substitusjoner inradically forskjellige måter.
pam matriser for aminosyrer, sammen med enkeltbokstaverbrukes til genetisk kodede aminosyrer, ble utviklet Av MargaretDayhoff. De ble opprinnelig publisert i 1978, og basert På proteinsekvensene Dayhoff hadde kompilert siden 1960-tallet, publisert som theAtlas Of Protein Sequence and Structure.
navnet PAM kommer fra «punkt akseptert mutasjon», og refererer tilplassering av en enkelt aminosyre i et protein med en annen aminosyre.Disse mutasjonene ble identifisert ved å sammenligne svært like sekvenser med minst 85% identitet, og det antas at eventuelle observerte substitusjoner var resultatet av en enkelt mutasjon mellom forfedresekvensen og en av dagens sekvenser.
PAM definerer også en tidsenhet, hvor 1 PAM er tiden der 1/100 aminosyrer forventes å gjennomgå en mutasjon. PAM1 sannsynlighet matrise showsthe sannsynligheten for aminosyren ved kolonne j blir erstattet av aminosyren på rad i. Det ble beregnet Fra Dayhoff ER PAM teller, og rescaled tobe 1 PAM tidsenhet. Som du kan se, er de diagonale sannsynlighetene ipam1-matrisen alle svært små (alle elementene ble skalert med 10.000 forlegibility):
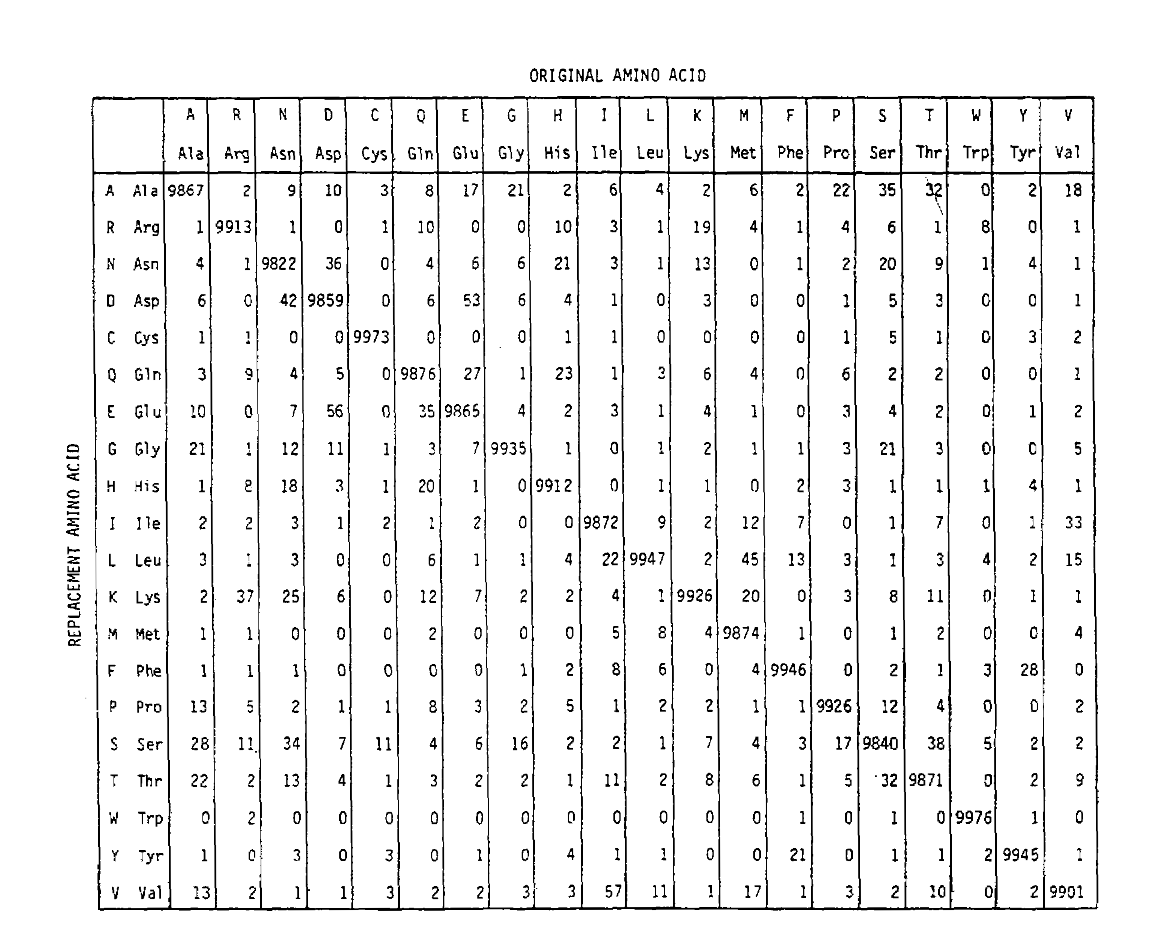
for å beregne aminosyreutskiftningssannsynlighetene for lengre tidsurasjoner, kan matrisen multipliseres med seg selv den tilsvarendeantall ganger. DERMED BLE PAM250-sannsynlighetsmatrisen, som beskriver sannsynligheter gitt 250 pam-tidsenheter, avledet ved å øke PAM1-sannsynlighetsmatrisen til kraften 250 (alle elementene ble skalert med 100 for lesbarhet):
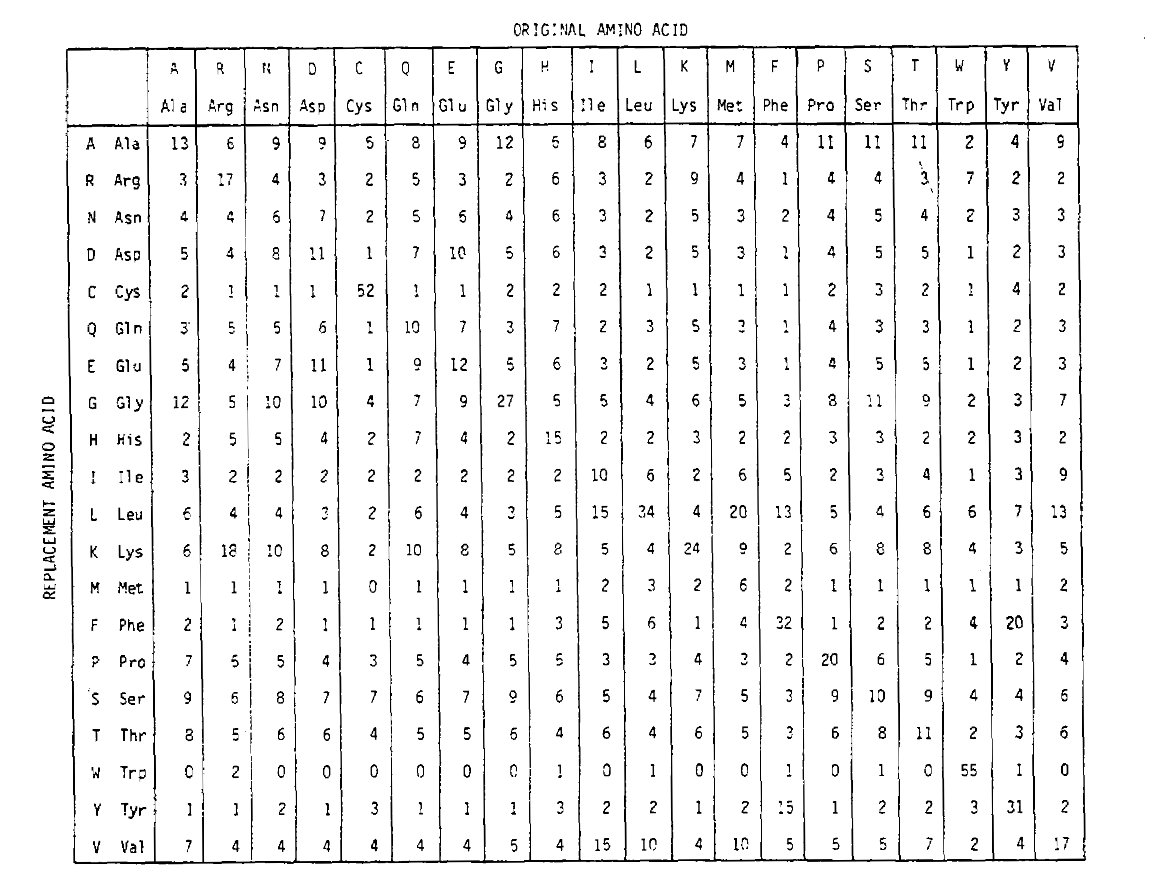
erstatningssannsynlighetene avledet ved hjelp av denne eksponeringen korrektkonto for flere substitusjoner. Ikke bare er off-diagonalproabiliteter proporsjonalt større som du forventer i lengre tidvarighet, men de er flattere. For eksempel er sannsynligheten for en erstatning med valin (V)til isoleucin (I) 33× større enn En Erstatning Med V til histadin (H)i PAM1-matrisen, men bare 4,5× større i PAM250-matrisen.
Score matriser kan da beregnes ut fra sannsynlighetsmatriser ogobserverte basefrekvenser.
BLOSUM matriser, utviklet Av Steven Og Jorja Henikoff og publisert i 1992, tar en helt annen tilnærming. MENS PAM implisitt anvender en stasjonær endelig nettstedsmodell for evolusjon ved hjelp av matriseutvidelse, behandles effekten av flere substitusjoner implisitt i BLOSUM byconstructing different score matriser for forskjellige tidsskalaer.
innenfor flere sekvensjusteringer av homologe sekvenser, bevarettilsvarende blokker av aminosyrer identifiseres. Innenfor hver blokk grupperes multiplesequences når deres parvise gjennomsnittlige sekvensidentitet er høyere enn noen terskel. Terskelen er 80% FOR blosum80-matrisen, 62% FOR BLOSUM62, 50% FOR BLOSUM50 og så videre.
dette betyr AT for BLOSUM80 vil blokker ha gjennomsnittlig parvis identitiesnei større enn 80%, FOR BLOSUM62 ikke større enn 62%, og så videre.
Aminosyreutskiftningssannsynligheter for homologe sekvenser beregnes fra parvise sammenligninger mellom klynger. Disse sannsynlighetene vil være resultatet av enkle og flere substitusjoner, med flere substitutionshaving større innflytelse på større evolusjonære avstander. Derfor vil scorematrices generert fra parvise sammenligninger mellom klynger av i gjennomsnitt større avstand, som BLOSUM50-matrisen, naturlig utgjøre den større effekten av flere substitusjoner.
Selv om de tar forskjellige ruter, den endelige BLOSUM og PAM score matricesare faktisk ganske like. Ifølge Henikoff og Henikoff er følgendepam og BLOSUM matriser sammenlignbare:
| PAM | BLOSUM |
|---|---|
| PAM250 | BLOSUM45 |
| PAM160 | BLOSUM62 |
| PAM120 | BLOSUM80 |
For more information on PAM (Dayhoff) and BLOSUM matrices, see kapittel 2 avbiologisk sekvensanalyse Av Durbin et al. Og Wikipedia.
Oppdatering 13. oktober 2019: For et annet perspektiv på substitusjonsmatriser, se avsnittet «Omveier» på slutten Av Kapittel 5 I Bioinformatikkalgoritmer (2. Eller 3. Utgave) Av Compeau og Pevzner.