o objetivo deste tutorial é apresentá-lo ao processamento de dados de sequenciação de próxima geração na galáxia. Este tutorial usa uma variante COVID-19 chamada a partir de dados ilumina, mas não é sobre a variante chamada per se.
na conclusão deste tutorial você saberá:
- Como encontrar dados em SRA e transferir esta informação para Galaxy
- Como executar o processamento básico de dados NGS no Galaxy, incluindo:
- Controle de Qualidade (QC) de Illumina de dados
- Mapeamento
- Remoção de duplicatas
- Variante chamada com
lofreq - Variante de anotação
- Usando conjuntos de dados de coleções
- Importação de dados para Jupyter
### Agenda>> neste tutorial, iremos abordar:>> 1. Cot > {: toc}>{: .agenda}# # dois caminhos através deste tutorialWe criou duas trajectórias que você pode seguir através deste tutorial.1. ** Trajetória 1 * * – Iniciar com a SRA do NCBI e buscar as adesões disponíveis → iniciar (#a sequência-leitura-Arquivo)2. ** Trajetória 2 * * – Bypass NCBI’s SRA and start with Galaxy directly. → Start (#back-in-galaxy)we recommending with **Trajectory 2**.# A sequência lida ArchiveThe (https://www.ncbi.nlm.nih.gov/sra) é o arquivo primário de *leituras não montadas* para o (https://www.ncbi.nlm.nih.gov/). A SRA é um ótimo lugar para obter os dados de sequenciamento que subjazem publicações e estudos.Este tutorial cobre como obter dados de sequência de SRA para a galáxia usando uma conexão direta entre os dois.> ### comment Comentário>> Você também vai ouvir a SRA referido como o *Curto Ler Arquivo*, seu nome original.> {:.comment}# # Accessing SRASRA can be reached either directly through it’s website, or through the tool panel on Galaxy.> ### comment Comentário>> Inicialmente, a ferramenta do painel de opção para acessar a SRA existe somente no (https://usegalaxy.org/). O suporte para a conexão direta com a SRA será incluído na versão 20.05 do Galaxy {:.comentário}> ### hands_on Hands-on: Explorar SRA Entrez>> 1. Vá para o seu Galaxy instância de escolha como um dos (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) ou qualquer outro. (Este tutorial usa usegalaxy.org).> 1. Se o seu histórico ainda não estiver vazio, então inicie um novo histórico (veja (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) para mais em histórias de galáxias)> 1. ** Clique em * * ‘Obter Dados’ no topo do painel de ferramentas.> 1. ** Clique em * * ‘SRA Server `na lista de ferramentas mostradas em`Get Data’.>this takes you the (https://www.ncbi.nlm.nih.gov/sra) — you can also start directly from the SRA. Uma caixa de pesquisa é mostrada no topo da página. Tente procurar por algo que lhe interesse, como` dolphin `ou` kidney `ou` Dolphin kidney `e depois **clique** o botão` Search’.>> isto devolve uma lista de experiências *SRA* que correspondem ao seu texto de pesquisa. Os experimentos SRA, também conhecidos como *entradas SRX*, contêm dados de sequência de um experimento particular, bem como uma explicação do experimento em si e quaisquer outros dados relacionados. Você pode explorar as experiências retornadas clicando em seu nome. Ver (https://www.ncbi.nlm.nih.gov/books/NBK56913/) no (https://www.ncbi.nlm.nih.gov/books/n/helpsrakb/) para mais.>> Quando estiver a introduzir texto na SRA caixa de pesquisa, você está usando (https://www.ncbi.nlm.nih.gov/sra/docs/srasearch/). Entrez suporta pesquisas de texto simples, e pesquisas muito precisas que verificam metadados específicos e usam expressões lógicas arbitrariamente complexas. O Entrez permite-lhe aumentar as suas pesquisas do básico para o avançado à medida que reduz as suas pesquisas. A sintaxe das pesquisas avançadas pode parecer assustadora, mas a SRA fornece uma gráfica (https://www.ncbi.nlm.nih.gov/sra/advanced/) para gerar a sintaxe específica. E, como veremos abaixo, o Selector de execução da SRA fornece uma interface de utilizador ainda mais amigável para estreitar os nossos dados seleccionados.>> Brincar com a SRA Entrez interface, incluindo o avançado construtor de consulta, para ver se você pode identificar um conjunto de SRA experiências que são relevantes para uma das suas áreas de investigação.{: .hands_on}> ### hands_on Hands-on: Gerar lista de correspondência de experimentos usando Entrez>> Agora que você tem uma familiaridade básica com a SRA Entrez, vamos encontrar as sequências usado neste tutorial.>> 1. Se você ainda não estiver lá, * * navegar * * de volta para o (https://www.ncbi.nlm.nih.gov/sra> 1. ** Limpar * * qualquer texto de pesquisa da caixa de pesquisa.> 1. ** Digite * * ‘sars-cov-2 `na caixa de pesquisa e **clique**`Search’.> isto devolve uma lista longa de experiências da SRA que correspondem à nossa pesquisa, e essa lista é demasiado longa para ser usada num exercício tutorial. Neste momento, poderíamos usar o avançado construtor de consultas Entrez que aprendemos sobre acima.> But we won’t. Instead lets send the *too long for a tutorial* list results we have to the SRA Run Selector, and use its friendlier interface to narrow our results.>> !(../../ images / sar_entrez.Forum){: .hands_on}> ### hands_on Hands-on: Go from Entrez to SRA Run Selector>> View results as an expanded interactive table using the RunSelector.>> 1. Carregue em enviar os resultados para executar o selector, que aparece numa caixa no topo dos resultados da pesquisa.>> !(../../ images / sra_ entrez_result.png)>>> # # tip What if you don’t see the Run Selector Link?>>>> Você pode ter notado esse texto antes, quando você estava explorando Entrez de pesquisa. Este texto só aparece algumas vezes, quando o número de resultados de pesquisa cai dentro de uma janela bastante larga. Você não vai vê-lo se você só tem alguns resultados, e você não vai vê-lo se você tem mais resultados do que o selector de execução pode aceitar.>>>> *Você precisa para começar a Executar o Seletor para enviar seus resultados para o Galaxy.* E se você não tiver resultados suficientes para ativar este link que está sendo mostrado? Nesse caso, ligue para o Selector de execução, carregando em* * * no menu ‘ Enviar para` no canto superior direito do painel de resultados. Para começar a executar o Selector, * * seleccione* * ‘Executar o Selector’ e depois** carregue em * * no botão `Ir`.> !(../../ images/sra_ entrez_send_to.png)> {: .dica}>>> 1. ** Clique em * * ‘Enviar resultados para executar o selector’ no topo do painel de resultados de pesquisa. (Se você não vê este link, então veja o comentário diretamente acima.){: .hands_on}# # SRA Run SelectorWe learned earlier how to narrow our search results by using Entrz’s advanced syntax. No entanto, não nos aproveitámos desse poder quando estávamos em Entrez. Em vez disso, usamos uma pesquisa simples e depois enviamos todos os resultados para o Selector de execução. Ainda não temos a (curta) lista de resultados que queremos analisar. O que estamos a fazer?* Estamos usando Entrez e o seletor de execução como eles são projetados para serem usados: * usar a interface Entrez para reduzir seus resultados para um tamanho que o seletor de execução pode consumir. * Enviar esses resultados Entrez para o seletor de execução SRA * usar a interface muito mais amigável do seletor de execução para 1. Compreender mais facilmente os dados que temos 1. Reduza esses resultados usando esse conhecimento.> # # # Selector de execução de comentários é tanto mais quanto menos do que Entrez>> O Selector de execução pode fazer mais, mas não tudo o que a sintaxe de busca de Entrez pode fazer. O selector de execução usa a tecnologia* faceted search * que é fácil de usar, e poderosa, mas que tem limites inerentes. Especificamente, o Entrez vai trabalhar melhor quando procurar em atributos que têm dezenas, centenas ou milhares de valores diferentes. O Selector de execução irá funcionar melhor na procura de atributos com menos de 20 valores diferentes. Felizmente, isso descreve a maioria das buscas.{: .comentário}a janela do Selector de execução está dividida em vários painéis:* **`lista de filtros`**: no canto superior esquerdo. É aqui que vamos refinar a nossa busca.* * * ` Selecione’**: um resumo do que foi inicialmente passado para executar o Seletor, e quanto do que selecionamos até agora. (E até agora, nós não selecionamos nada disso.) Notem também o tentador, mas ainda tingido, botão `Galaxy`.** * ` Found x Items ‘ * * inicialmente, esta é a lista de itens enviados para executar o Selector de Entrez. Esta lista irá encolher à medida que aplicamos filtros a ela.!(../../ images/sra_ run_selector.png)> # # # comment Why did the number of found items * go up?* >> Recall that the Entrez interface lists SRA experiments (SRX entries). Run Selector lists * runs * — sequencing datasets-and there are* one or more * runs per experiment. Temos os mesmos dados de antes, estamos agora a vê-los em mais pormenor.{: .comentário}a ‘lista de filtros’ no canto superior esquerdo mostra colunas nos nossos resultados que têm valores numéricos contínuos ou 10 ou menos (pode alterar este número) valores distintos neles. ** Role* * para baixo através da lista selecione alguns dos filtros. Quando um filtro é selecionado, uma caixa de *valores* aparece abaixo, listando as opções para este filtro, e o número de corridas com cada opção. Estes valores / opções são extraídos dos metadados do conjunto de dados. Tente * * * seleccionar * alguns filtros de som interessantes e depois** Seleccionar * * uma ou mais opções para cada filtro. Tente * * deseleccionar * * opções e filtros. Como você faz isso, o número de Resultados encontrados vai diminuir ou aumentar.> # # tip Tip: Utilize os Filtros para melhor entender os dados de>> Filtros são como você a restringir os conjuntos de dados sob consideração para o envio da Galáxia, mas eles também são uma excelente forma de compreender os seus dados:> em Primeiro lugar, seleccionar um filtro, é uma maneira fácil para ver a gama de valores em uma coluna. Você pode não ser capaz de (https://www.google.com/search?q=sra+sirs_outcome), mas você pode possivelmente descobri-lo vendo que valores estão nele.> Second, you can explore how different columns relate to each other. Existe uma relação entre os valores “sirs_outcome” e os valores “disease_stage”?{: .dica}> ### hands_on Hands-on: restringir os resultados a utilização de Executar o Selector>> 1. Se tiver algum filtro ligado, * * deseleccione * * eles.> uma vez que você tenha feito isso, não haverá quaisquer *valores* caixas aparecendo abaixo da `lista de filtros`.> 2. ** Copiar e colar * * este texto de pesquisa no campo de pesquisa ‘itens encontrados’.>> SRR11772204 OU SRR11597145 OU SRR11667145>> Esta escolhidos a dedo conjunto de execuções limites de nossos resultados para 3 é executado a partir de diferentes região de distribuição geográfica.{: .hands_ on}isto reduz a sua lista de ‘itens encontrados’ de dezenas de milhares de runs para 3 runs (um número controlável para um tutorial!). Mas ainda não acabámos o selector de corrida. Note que o botão ‘Galaxy’ ainda está acinzentado. Reduzimos as nossas opções, mas ainda não seleccionámos nada para enviar para a galáxia.É possível seleccionar todos os restantes executados por * * Click* * o marcador no topo da primeira coluna. Pode deseleccionar tudo se carregar em* * * * no ‘X’.> ### hands_on Hands-on: Selecione executa e enviar para o Galaxy>> 1. Seleccione Todas as corridas com * * clicando* * o ‘X’.> And now, the` Galaxy ‘ button is live.> 1. ** Clique em * * o botão ‘Galaxy` na seção ‘Select’ no topo da página.{: .hands_on} # # de volta à galáxia quando clicamos em ‘ Galaxy` no Selector de execução várias coisas acontecem. Primeiro, ele lança uma nova página de navegador ou janela que se abre no Galaxy. Você vai ver a caixa * grande verde * indicando que o aperto de mão entre a SRA e o Galaxy foi bem sucedido e então você vai ver um novo trabalho `SRA` em seu painel de história. Esta caixa pode começar como cinzenta / pendente, indicando que a transferência ainda não começou, ou pode ir directamente para o amarelo / em execução ou para o verde / feito.> # # hands_on Hands-on: Examine o novo conjunto de dados SRA>> 1. Uma vez que a transferência` SRA ‘ esteja completa, **clique** no ícone galáxia-olho do dataset (eye).>> isto mostra o conjunto de dados no painel central do Galaxy.{: .hands_on}o conjunto de dados` SRA ‘ não é dados de sequência, mas sim metadados *que vamos usar para obter dados de sequência da SRA. Estes meta-dados espelham a informação que vimos na secção “Itens encontrados” do selector de execução. Os metadados não são os dados finais que estamos buscando da SRA, mas ter todos esses metadados é muitas vezes útil em etapas de análise subsequentes.Vamos agora usar esses meta-dados para obter os dados da sequência da SRA. A SRA fornece ferramentas para extrair todo tipo de informação, incluindo os próprios dados de sequência. A ferramenta Galaxy ‘Faster Download and Extract Reads in FASTQ’ é baseada no utilitário SRA (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump), e faz exatamente isso.– >
- Encontre os dados necessários em SRA
- hands_on Hands-on: descrição da Tarefa
- comment Comment
- processo e filtro SraRunInfo.csv file in Galaxy
- hands_on Hands-on: Upload SraRunInfo.ficheiro csv no Galaxy
- comment Beware of Cuts
- hands_on Hands-on: Creating a subset of data
- tip Tip Tip: Finding tools
- Download de dados de sequenciamento com mais Rápido de Baixar e Extrair Lê em FASTQ
- hands_on Hands-on: descrição da Tarefa
- Análise de variação dos dados de sequenciação SARS-Cov-2
- comente a usegalaxia.* COVID-19 analysis project
- obter os dados do genoma de referência
- hands_on Hands-on: Get the reference genome data
- Sugestão: a Importação através de links
- Adaptador de desbaste com fastp
- hands_on Hands-on: descrição da Tarefa
- Alinhamento com o Mapa com a BWA-MEM
- hands_on Hands-on: Alinhar as leituras sequenciais com o genoma de referência
- Remover duplicatas com MarkDuplicates
- hands_on Hands-on: Remove PCR duplicates
- Gerar alinhamento estatísticas com Samtools estatísticas
- hands_on Hands-on: Gerar alinhamento estatísticas
- Realinhar lê com lofreq viterbi
- hands_on Hands-on: Realinhar lê em torno de indels
- Adicionar ao qualidades com lofreq Inserir indel qualidades
- hands_on Hands-on: Adicionar ao qualidades
- Chamadas Variantes usando lofreq Chamada variantes
- hands_on Hands-on: Chamada de variantes
- anotar efeitos variantes com efeito SnpEff:
- hands_on Hands-on: annotate variant effects
- crie uma tabela de variantes usando campos de extrato de SnpSift
- hands_on Hands-on: Criar tabela de variantes
- resuma dados com MultiQC
- hands_on Hands-on: Resumir dados
- Conclusão
- keypoints pontos-Chave
- Perguntas mais Frequentes
- literatura Útil
- Citando este Tutorial
- details BibTeX
Encontre os dados necessários em SRA
primeiro precisamos encontrar um bom conjunto de dados para brincar. O Sequence Read Archive (SRA) é o principal arquivo de leituras não montadas operadas pelos Institutos Nacionais de saúde dos Estados Unidos (NIH). A SRA é um ótimo lugar para obter os dados de sequenciamento que subjazem publicações e estudos. Vamos fazer o seguinte:
hands_on Hands-on: descrição da Tarefa
- Ir para NCBI é a SRA página apontando seu navegador para https://www.ncbi.nlm.nih.gov/sra
- Na caixa de pesquisa, insira
SARS-CoV-2 Patient Sequencing From Partners / MGH(Alternativamente, você simplesmente clicar neste link)
- a página web irá mostrar um grande número de conjuntos de dados da SRA (no momento da escrita havia 2.223). Estes são dados de um estudo que descreve a análise da SARS-CoV – 2 na área de Boston.
- Download metadados que descrevem estes conjuntos de dados:
- clicar em Enviar para: pendente
- Seleccionar
File- Alterar Formato para
RunInfo- Clicando em Criar fileHere é como ele deve se parecer com:
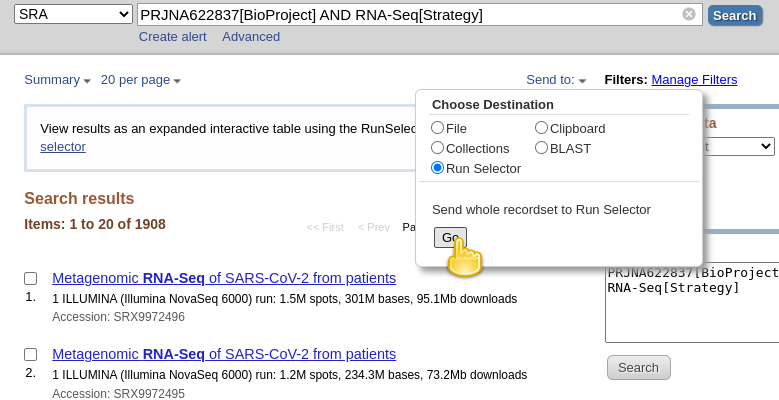
- isto criaria uma grande
SraRunInfo.csvficheiro na suaDownloadspasta.
Agora que baixámos este ficheiro, podemos ir para uma instância Galaxy e começar a processá-lo.
comment Comment
Note que o ficheiro que acabámos de descarregar não está a sequenciar os dados em si. Em vez disso, são metadados descrevendo propriedades de leitura sequenciada. Vamos filtrar esta lista para apenas algumas adesões que serão usadas no restante deste tutorial.
processo e filtro SraRunInfo.csv file in Galaxy
hands_on Hands-on: Upload SraRunInfo.ficheiro csv no Galaxy
- vá à sua instância de escolha da galáxia, tal como uma das usegalaxy.org, usegalaxy.eu, usegalaxy.org.au ou qualquer outro. (Este tutorial usa usegalaxy.org).
- carregue no botão enviar dados:
- Na caixa de diálogo que será exibida clique “Escolher arquivos locais” botão:

- selecione
SraRunInfo.csvarquivo do seu computador- Clique no botão Iniciar
- Fechar a caixa de diálogo pressionando o botão Fechar
- Você pode agora examinar o conteúdo deste arquivo clicando galaxy-eye (olho) ícone. Você verá que este arquivo contém muitas informações sobre adesões individuais da SRA. Neste estudo, cada adesão corresponde a um doente individual cujas amostras foram sequenciadas.
Galaxy can process all 2,000+ datasets but to make this tutorial bearable we need to selected a smaller subset. Em particular, a nossa experiência anterior com estes dados mostra dois conjuntos de dados interessantes SRR11954102 e SRR12733957. Então, vamos tirá-los.
comment Beware of Cuts
The Hands-on section below uses Cut tool. Existem duas ferramentas cortadas na galáxia devido a razões históricas. Este exemplo usa a ferramenta com o nome completo Cut colunas de uma tabela (cut). No entanto, a mesma lógica se aplica à outra ferramenta. Ele simplesmente tem uma interface ligeiramente diferente.
hands_on Hands-on: Creating a subset of data
- Find tool” Select lines that match an expression ” tool in Filter and Sort section of the tool panel.
tip Tip Tip: Finding tools
Galaxy may have an esmagador amount of tools installed. Para encontrar uma ferramenta específica digite o nome da ferramenta na caixa de pesquisa do painel de ferramentas para encontrar a ferramenta.
- certifique-se de que o
SraRunInfo.csvdataset que acabamos de enviar está listado no arquivo param” selecione linhas “do campo” da forma de Ferramenta.- in” the pattern ” field enter the following expression →
SRR12733957|SRR11954102. Estas são duas adesões que queremos encontrar separadas pelo símbolo do tubo||significaor: localizar linhas que contêmSRR12733957ouSRR11954102.- clique em
Executebotão.- isto irá gerar um ficheiro que contém duas linhas (bem … uma linha também é usada como cabeçalho, por isso irá aparecer que o ficheiro tem três linhas. Está tudo bem.)
- corta a primeira coluna do ficheiro usando a ferramenta “Cortar”, que irá encontrar na secção de manipulação de texto da área de ferramentas.
- certifique-se que o conjunto de dados produzido pela etapa anterior é seleccionado no campo “ficheiro a cortar” do formulário de Ferramenta.
- Change “Delimited by” to
Comma- In “List of fields” select
Column: 1.- Tecle
ExecuteIsso irá produzir um arquivo de texto com apenas duas linhas:SRR12733957SRR11954102
Agora que temos identificadores de conjuntos de dados que queremos precisamos de baixar o real de dados de sequenciamento.
Download de dados de sequenciamento com mais Rápido de Baixar e Extrair Lê em FASTQ
hands_on Hands-on: descrição da Tarefa
- mais Rápido de Baixar e Extrair Lê em FASTQ ferramenta com os seguintes parâmetros:
- “selecione o tipo de entrada”:
- o parâmetro param-file “sra accession list” deve apontar o resultado da ferramenta “Cut” do passo anterior.
- clique no botão
Execute. Isto irá executar a ferramenta, que recupera a sequência ler conjuntos de dados para as corridas que foram listadas no conjunto de dadosSRAdataset. Pode levar algum tempo. Então esta pode ser uma boa hora para fazer o café.- várias entradas são criadas no seu painel de história quando enviar esta tarefa:
Pair-end data (fasterq-dump): Contém Emparelhado fim de conjuntos de dados (se disponível)Single-end data (fasterq-dump)Contém Único fim de conjuntos de dados (se disponível)Other data (fasterq-dump)Contém Ímpar conjuntos de dados (se disponível)fasterq-dump logContém Informações sobre a ferramenta de execução
Os três primeiros itens são, na verdade, conjuntos de conjuntos de dados. Coleções na Galáxia são agrupamentos lógicos de conjuntos de dados que refletem as relações semânticas entre eles no experimento / análise. Neste caso, a ferramenta cria uma coleção separada para leituras emparelhadas, Leituras simples, e outras.Veja os tutoriais das coleções para mais informações.
Explore as colecções Carregando primeiro no nome da colecção no painel Histórico. Isto leva-o para dentro da colecção e mostra-lhe os conjuntos de dados nela. Você pode então navegar de volta para o nível exterior de sua história.
Uma vez fasterq termina a transferência de dados (todas as caixas são verdes / feitas), estamos prontos para analisá-lo.e agora?
você pode agora analisar os dados recuperados usando qualquer ferramenta de análise de seqüências e fluxos de trabalho na galáxia. A SRA detém dados de suporte para cada tipo de experimento *-seq imaginável.
se executou este tutorial, mas obteve conjuntos de dados em que estava interessado, então veja o resto da biblioteca GTN para obter ideias sobre como analisar na galáxia.
no entanto, se você recuperou os conjuntos de dados usados nos exemplos deste tutorial acima, então você está pronto para executar a análise da variante SARS-CoV – 2 abaixo.
Análise de variação dos dados de sequenciação SARS-Cov-2
nesta parte do tutorial vamos realizar chamada variante e análise básica dos conjuntos de dados baixados acima. Vamos começar por baixar a sequência de referência Wuhan-Hu-1 SARS-CoV-2, em seguida, executar a remoção do adaptador, alinhamento e chamada variante e, finalmente, olhar para a distribuição geográfica de algumas das variantes encontradas.
comente a usegalaxia.* COVID-19 analysis project
This tutorial uses a subset of the data and runs through thevariation Analysissection of covid19.galaxyproject.organizacao.Os dados para covid19.galaxyproject.org está a ser actualizado continuamente à medida que novos conjuntos de dados são tornados públicos.
obter os dados do genoma de referência
os dados do genoma de referência para hoje são para SARS-CoV-2, “síndrome respiratória aguda grave coronavírus 2 isolado Wuhan-Hu-1, genoma completo”, Tendo o ID de adesão de NC_045512.2.
estes dados estão disponíveis a partir do Zenodo usando a seguinte ligação.
hands_on Hands-on: Get the reference genome data
Import the following file into your history:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzSugestão: a Importação através de links
- Copiar a localização do link
- Abra o Galaxy Carregar Gerenciador (galaxy-upload no canto superior direito do painel de ferramentas)
- Selecione Colar/Obtenção de Dados
- Cole o link no campo de texto
- Pressione Start
- Feche a windowBy padrão, o Galaxy usa a URL como o nome, portanto, mude o nome os ficheiros com um nome mais útil.
Adaptador de desbaste com fastp
seqüenciamento Remoção de adaptadores de melhora alinhamentos e variante chamada. a ferramenta fastp pode detectar automaticamente adaptadores de sequenciamento amplamente utilizados.
hands_on Hands-on: descrição da Tarefa
- fastp ferramenta com os seguintes parâmetros:
- “Single-end ou emparelhado lê”:
Paired Collection
- param-ficheiro”, Seleccione emparelhado coleção(s)”:
list_paired(saída de mais Rápido de Baixar e Extrair Lê em FASTQ ferramenta)- Em “Opções de Saída”:
- “Saída JSON relatório”:
Yes
Alinhamento com o Mapa com a BWA-MEM
BWA-MEM é uma ferramenta amplamente utilizada seqüência aligner para curto-leitura de sequenciamento de conjuntos de dados tais como os que estamos analisando neste tutorial.
hands_on Hands-on: Alinhar as leituras sequenciais com o genoma de referência
- mapa com a ferramenta BWA-MEM com os seguintes parâmetros:
- “irá seleccionar um genoma de referência da sua história ou usar um índice incorporado?”:
Use a genome from history and build index
- param-arquivo “Usar o seguinte conjunto de dados como referência sequência”:
output(conjunto de dados de Entrada)- “Único ou Emparelhado-end lê”:
Paired Collection
- param-ficheiro”, Seleccione um par de sites”:
output_paired_coll(saída de fastp ferramenta)- “Conjunto de ler grupos de informações?”:
Do not set- “Selecionar” modo de análise:
1.Simple Illumina mode
Remover duplicatas com MarkDuplicates
MarkDuplicates ferramenta remove duplicate sequências de origem a partir de uma biblioteca de preparação de artefatos de seqüenciamento e de artefatos. É importante remover estas sequências de artefacto para evitar uma sobre-representação artificial de uma única molécula.
hands_on Hands-on: Remove PCR duplicates
- MarkDuplicates tool with the following parameters:
- param-ficheiro”, Seleccione SAM/BAM conjunto de dados ou conjunto de dados de coleta”:
bam_output(saída do Mapa com a BWA-MEM ferramenta)- “Se for verdadeiro não escreva duplicatas para o arquivo de saída, em vez de escrevê-las com sinalizadores apropriados set”:
Yes
Gerar alinhamento estatísticas com Samtools estatísticas
Após a marcação duplicada passo acima, podemos gerar estatística sobre o alinhamento temos gerado.
hands_on Hands-on: Gerar alinhamento estatísticas
- Samtools estatísticas de ferramenta com os seguintes parâmetros:
- param-arquivo “BAM arquivo”:
outFile(saída de MarkDuplicates ferramenta)- “Definir a cobertura de distribuição”:
No- “Saída”:
One single summary file- “Filtrar por SAM bandeiras”:
Do not filter- “Usar uma referência sequência”:
No- “Filtrar por regiões”:
No
Realinhar lê com lofreq viterbi
Realinhar lê ferramenta corrige os desvios em torno de inserções e exclusões. Isto é necessário para detectar com precisão variantes.
hands_on Hands-on: Realinhar lê em torno de indels
- Realinhar lê com lofreq ferramenta com os seguintes parâmetros:
- param de-ficheiro “, Lê-se para realinhar”:
outFile(saída de MarkDuplicates ferramenta)- “, Escolher a fonte para a referência de genoma”:
History
- param-arquivo “Reference”:
output(conjunto de dados de Entrada)- Em “opções Avançadas”:
- “Como lidar com base qualidades de 2?”:
Keep unchanged
Adicionar ao qualidades com lofreq Inserir indel qualidades
Este passo adiciona ao qualidades para a nossa ficheiro do alinhamento. Isto é necessário para chamar variantes usando variantes de chamada com lofreq tool
hands_on Hands-on: Adicionar ao qualidades
- Insira a indel qualidades com lofreq ferramenta com os seguintes parâmetros:
- param de-ficheiro “, Lê-se”:
realigned(saída de Realinhar lê ferramenta)- “Indel cálculo abordagem”:
Dindel
- “, Escolher a fonte para a referência de genoma”:
History
- param-arquivo “Reference”:
output(conjunto de dados de Entrada)
Chamadas Variantes usando lofreq Chamada variantes
agora Estamos prontos para chamadas variantes.
hands_on Hands-on: Chamada de variantes
- Chamadas variantes com lofreq ferramenta com os seguintes parâmetros:
- param-file “Input lê em BAM” formato:
output(saída de Inserir a indel qualidades ferramenta)- “, Escolher a fonte para a referência de genoma”:
History
- param-arquivo “Reference”:
output(conjunto de dados de Entrada)- “Chamada variantes”:
Whole reference- “Tipos de variantes de chamar de”:
SNVs and indels- “Variante chamada” parâmetros:
Configure settings
- Na “Cobertura”:
- “o Mínimo de cobertura”:
50- Na “Base-calling”:
- “Mínimo baseq”:
30- “Mínimo baseq alternativo bases”:
30- no “mapeamento qualityy
20- “variante parâmetros de filtro”:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
a saída deste passo é uma coleção de arquivos VCF que podem ser visualizados em um navegador do genoma.
anotar efeitos variantes com efeito SnpEff:
vamos agora anotar as variantes que chamámos na etapa anterior com o efeito que têm no genoma SARS-CoV-2.
hands_on Hands-on: annotate variant effects
- SnpEff eff: tool with the following parameters:
- param-file ” Sequence changes (SNPs, MNPs, InDels)”:
variants(saída de Chamadas variantes ferramenta)- “formato de Saída”:
VCF (only if input is VCF)- “Criar relatório de CSV, útil para fins de análise (-csvStats)”:
Yes- “Anotação de opções”: `
- “Filtrar resultados”: `
- “Filtrar Efeitos específicos”:
No
O resultado deste passo é um arquivo VCF com adição de variante efeitos.
crie uma tabela de variantes usando campos de extrato de SnpSift
iremos agora selecionar vários efeitos do VCF e criar um arquivo tabular que é mais fácil de entender para os seres humanos.
hands_on Hands-on: Criar tabela de variantes
- SnpSift Campos Extrair ferramenta com os seguintes parâmetros:
- param-arquivo “Variante de entrada do arquivo no formato VCF”:
snpeff_output(saída de SnpEff fep: ferramenta)- os Campos “extrair”:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- “vários separador de campo”:
,- “campo vazio texto”:
.
podemos inspecionar os arquivos de saída e consulte verificar se as Variantes em que este ficheiro, também são descritas em um observável notebook que mostra a distribuição geográfica de SARS-CoV-2 variante sequências
Interessante variantes incluem o C a T variante na posição 14408 (14408C/T) em SRR11772204, 28144T/C em SRR11597145 e 25563G/T SRR11667145.
resuma dados com MultiQC
vamos agora resumir a nossa análise com MultiQC, que gera um belo relatório para os nossos dados.
hands_on Hands-on: Resumir dados
- MultiQC ferramenta com os seguintes parâmetros:
- Em “Resultados”:
- param repetir “Inserir Resultados”
- “, o Qual ferramenta foi usada gerar logs?”:
fastp
- param-file “Output of fastp”:
report_json(saída da ferramenta fastp)param-repetir “inserir Resultados”
- “que ferramenta foi usada gerar registos?”:
Samtools
- Em” saída de Samtools”:
- param-repeat” inserir saída de Samtools “
- ” tipo de saída de Samtools?”:
stats
- param-file “Samtools stats output”:
output(saída de Samtools estatísticas de ferramenta)- param repetir “Inserir Resultados”
- “, o Qual ferramenta foi usada gerar logs?”:
Picard
- In” Picard output”:
- param-repeat” Insert Picard output “
- ” Type of Picard output?”:
Markdups- param-file “Picard output”:
metrics_file(output of MarkDuplicates tool)param-repeat” Insert Results “
- ” Que ferramenta foi usada para gerar registos?”:
SnpEff
- param-arquivo de Saída”, de SnpEff”:
csvFile(saída de SnpEff fep: ferramenta)
Conclusão
Parabéns, agora você já sabe como importar dados de seqüência da SRA e como executar um exemplo de uma análise sobre esses conjuntos de dados.
keypoints pontos-Chave
a Sequência de dados em que a SRA pode ser diretamente importado para o Galaxy
Perguntas mais Frequentes
Tem dúvidas sobre este tutorial? Confira a página FAQ para o tópico de análise variante para ver se a sua pergunta está listada lá. Se não, por favor, faça sua pergunta sobre o GTN Gitter Canal ou o Galaxy Fórum de Ajuda
literatura Útil
Mais informações, incluindo links para a documentação e publicações originais, sobre as ferramentas, técnicas de análise e interpretação dos resultados descritos neste tutorial pode ser encontrado aqui.usou este material como instrutor? Sinta-se à vontade para nos dar feedback sobre como correu.
Citando este Tutorial
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 a Partir do NCBI Sequência de Ler Arquivo (SRA) da Galáxia: a SARS-CoV-2 variante de análise (Galaxy Materiais de Treinamento). / training-material / topics/variant-analysis/tutorials/sars-cov-2 / tutorial.html Online; accessed TODAY
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j. cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}