aminoácidos, nucleótidos ou qualquer outro caráter evolutivo são substituídos por outros a alguma taxa. Por exemplo, imagine uma seqüência evolucionária com threepossible estados, A, B e C. Se o processo de substituição do modelo é de tempo reversível, therewill ser três taxas de transição, Uma<>B, B<>C e Uma<>C.
suponha que as taxas sejam 1, 1 e 0, respectivamente, em unidades de substituição por 100 caracteres por unidade de tempo. Após uma unidade de tempo, em uma sequência de 300 caracteres originalmente composta igualmente de As, BS e Cs, esperamos que tenha havido uma substituição de A A B e uma substituição de B A C. Se estamos comparando duas sequências homólogas em organismos vivos, porque uma unidade de tempo passou para ambas as sequências, esperaríamos duas a A B e duas B A Csubstitutions entre as sequências atuais.
não importa o tempo que percorremos este processo, nunca haverá uma recolocação direta de A por C. Também nunca haverá uma substituição de A A C sob o modelo aso-chamado Infinite sites, onde não mais do que uma substituição pode ocorrer em um único site.
no entanto, uma vez que substituições A A B E B A C são comuns, sob um modelo finito B eventualmente será substituído por C em um local onde A foi anteriormente substituído por B. Esta substituição indireta de A por C (ou equivalentemente em um modelo reversível de tempo, C por a) torna-se mais provável quanto mais tempo os períodos de separação das sequências homólogas.
I simulou a evolução da sequência com base no cenário acima, rodando a simulação por 10 unidades de tempo. A partir desta substituição, observei as seguintes contagens para cada padrão de Sítio:
| A | B | C | |
|---|---|---|---|
| A | 91 | 9 | 0 |
| B | 5 | 86 | 9 |
| C | 0 | 9 | 91 |
neste duração relativamente curta, ele não aparece como se qualquer Um<>Csubstitutions ter ocorrido. No entanto, quando eu reanalisei a simulação por 100 unidades de tempo:
| A | B | C | |
|---|---|---|---|
| A | 55 | 35 | 10 |
| B | 29 | 36 | 35 |
| C | 20 | 36 | 44 |
Como você pode ver, muitos caracteres “A” foram substituídos por “C” e vice-versa. Mais genericamente, sob um modelo finito de sites múltiplas substituições porque a distribuição de padrões de site conta para se tornar muito mais lisonjeiro além simplesmente aumentando a proporção de off-diagonal em relação às contagens diagonais.As matrizes de pontuação PAM e BLOSUM são responsáveis por múltiplas substituições de maneiras diferentes.
As matrizes PAM para aminoácidos, juntamente com as abreviaturas de letras únicas usadas para aminoácidos geneticamente codificados, foram desenvolvidas por MargaretDayhoff. Eles foram originalmente publicados em 1978, e com base nas proteinsequências que Dayhoff tinha vindo a compilar desde a década de 1960, publicado como teatros de sequência e estrutura proteica.
o nome PAM vem de “mutação aceite ponto”, e refere-se à colocação de um único aminoácido numa proteína com um aminoácido diferente.Estas mutações foram identificadas através da comparação de sequências altamente semelhantes com pelo menos 85% de identidade, e assume-se que qualquer substituição observada foi o resultado de uma única mutação entre a sequência ancestral e uma das actuais sequências de dias.
PAM também define uma unidade de tempo, onde 1 PAM é o tempo em que se espera que 1/100 aminoácidos sofram uma mutação. A matriz de probabilidade PAM1 mostra a probabilidade de o aminoácido na coluna j ser substituído pelo aminoácido na linha I. foi calculada a partir das contagens PAM de Dayhoff, e redescobriu a unidade de tempo Tobe 1 PAM. Como você pode ver, o fora da diagonal probabilidades de thePAM1 matriz são todos muito pequenos (todos os elementos foram escalados por 10.000 forlegibility):
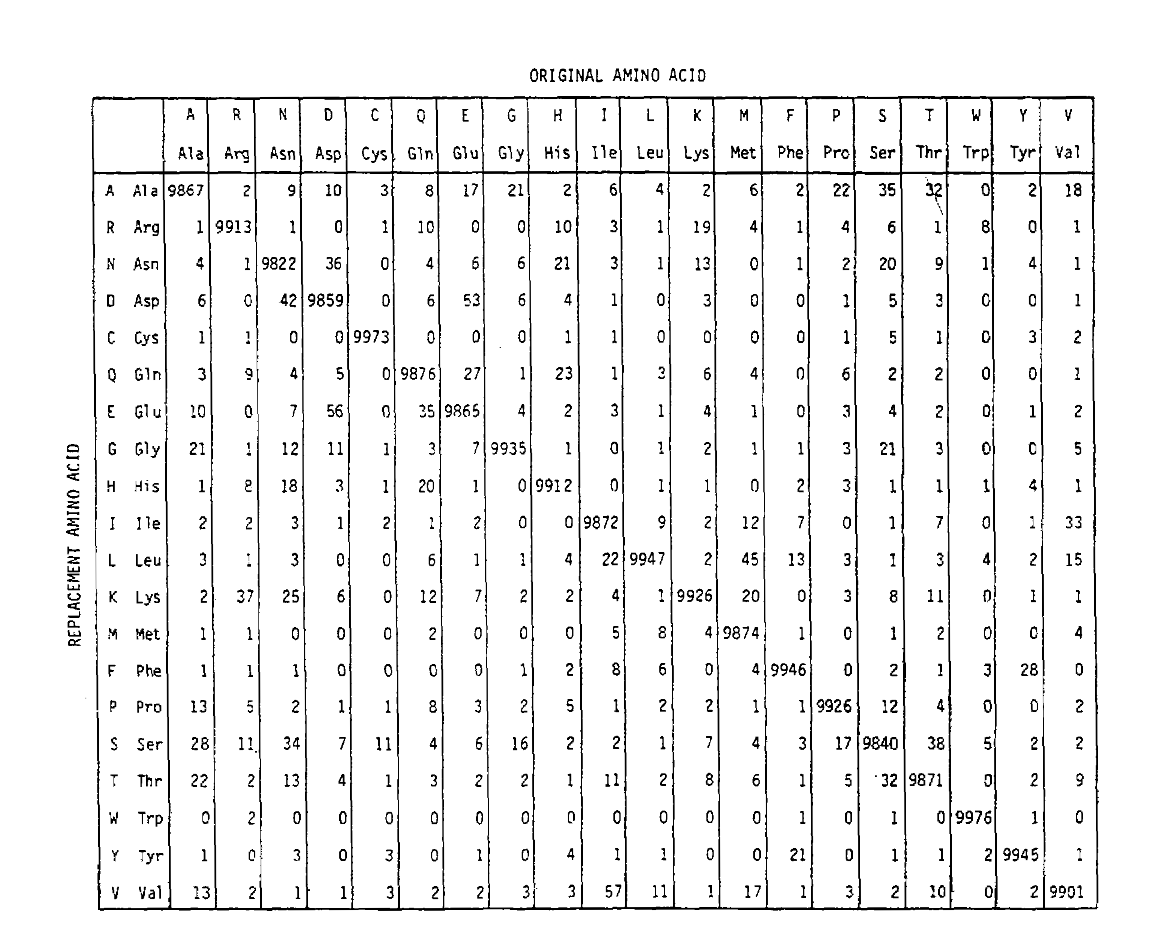
Para calcular o aminoácido de substituição de probabilidades para mais timedurations, a matriz pode ser multiplicado por si só o correspondingnumber de vezes. Assim, a matriz de probabilidade PAM250, descrevendo as probabilidades deplacement dadas 250 unidades de tempo PAM, foi derivada aumentando a matriz de probabilidade PAM1 para a potência 250 (todos os elementos foram escalados por 100 para legibilidade):
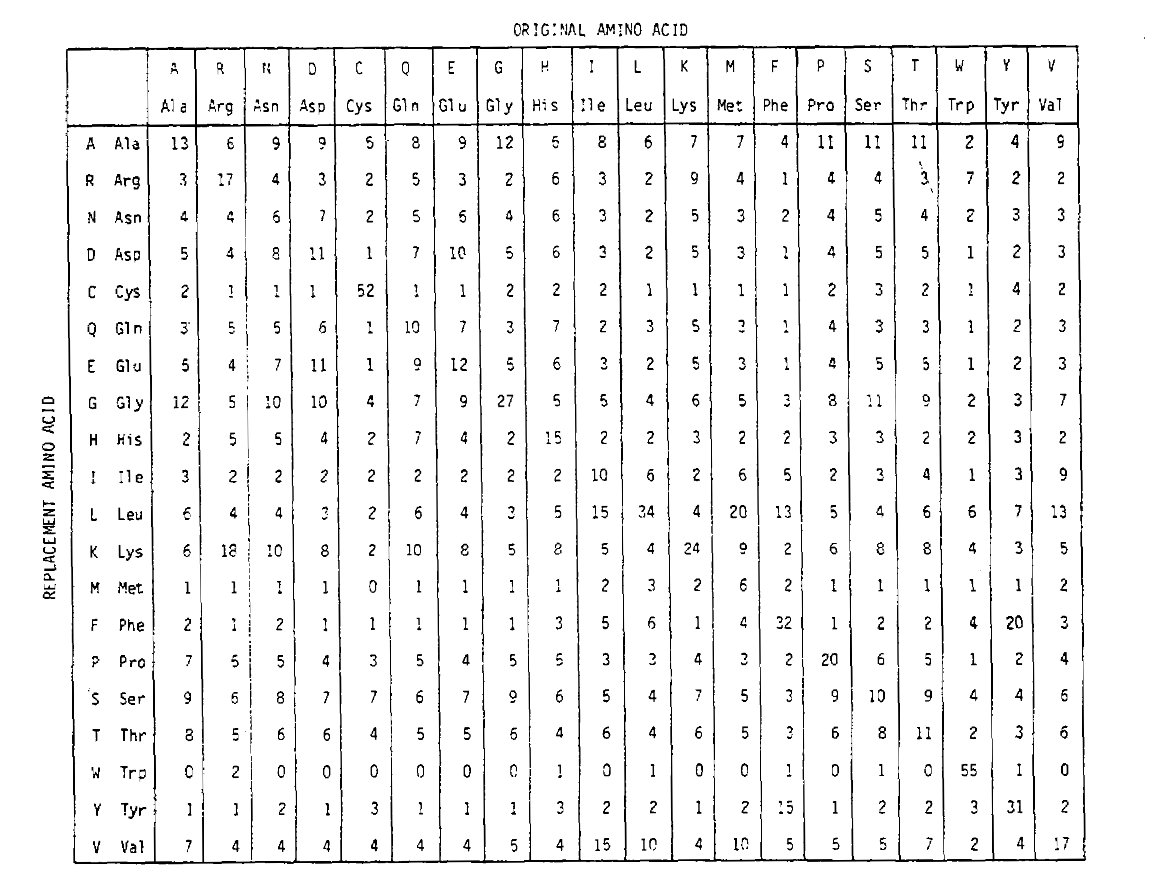
as probabilidades de substituição derivadas usando esta exponenciação correctlyaccount para substituições múltiplas. Não só as improbabilidades off-diagonal são proporcionalmente maiores como você esperaria para uma duração mais longa, mas eles são mais lisonjeiros. Por exemplo, a probabilidade de uma substituição de valina (V)para isoleucina (I) é 33× maior do que a substituição de um V para histadina (H)na matriz PAM1, mas apenas 4,5× maior na matriz PAM250.
matrizes de Pontuação podem então ser calculadas a partir das matrizes de probabilidade e frequências de base observadas.as matrizes BLOSUM, desenvolvidas por Steven e Jorja Henikoff e publicadas em 1992, têm uma abordagem muito diferente. Enquanto a PAM é implicitamente aplicada a um modelo fixo de locais finitos de evolução usando exponenciação de matriz, o efeito de múltiplas substituições é tratado implicitamente em BLOSUM por ter construído diferentes matrizes de pontuação para diferentes escalas de tempo.
dentro de alinhamentos de sequência múltipla de sequências homólogas, são identificados blocos contíguos de aminoácidos conservados. Dentro de cada bloco, multiplesequências são agrupadas quando sua identidade de sequência média emparelhada é maior do que algum limiar. O limiar é de 80% para a matriz BLOSUM80, 62% para BLOSUM62, 50% para BLOSUM50 e assim por diante.
isto significa que, para o BLOSUM80, os blocos terão identidadesparal média não superior a 80%, para o BLOSUM62 não superior a 62%, etc.
as probabilidades de substituição de aminoácidos para sequências homólogas são calculadas a partir de comparações emparelhadas entre grupos. Estas probabilidades serão o resultado de substituições simples e múltiplas, com substituições múltiplas que poupam maior influência a distâncias evolucionárias maiores. Por conseguinte, os índices de pontuação gerados a partir de comparações emparelhadas entre agregados de uma distância média maior, como a matriz BLOSUM50, irão naturalmente ter em conta o efeito mais elevado das substituições múltiplas.
embora eles tomem diferentes rotas, o BLOSUM final e PAM pontuação matrices são realmente muito semelhantes. De acordo com Henikoff e Henikoff, as matrizes followingPAM e BLOSUM são compreensíveis:
| PAM | BLOSUM |
|---|---|
| PAM250 | BLOSUM45 |
| PAM160 | BLOSUM62 |
| PAM120 | BLOSUM80 |
For more information on PAM (Dayhoff) and BLOSUM matrices, see chapter 2 ofBiological sequence analysis by Durbin et al., and Wikipedia.
Update 13 October 2019: for an another perspective on substitution matrices, consult the “Detours” section at the end of Chapter 5 of Bioinformatics Algorithms (2nd or 3rd Edition) by Compeau and Pevzner.