scopul acestui tutorial este de a vă prezenta procesarea datelor de secvențiere de generație următoare în Galaxy. Acest tutorial folosește o variantă COVID-19 apelând din datele Illumina, dar nu este vorba despre apelarea variantelor în sine.
la finalizarea acestui tutorial veți ști:
- cum să găsiți date în SRA și să transferați aceste informații către Galaxy
- cum să efectuați procesarea de bază a datelor NGS în Galaxy, inclusiv:
- controlul calității (QC) de date Illumina
- Mapping
- eliminarea duplicatelor
- varianta de asteptare cu
lofreq - varianta adnotare
- folosind colecții seturi de date
- importul de date la Jupyter
### Agenda>> în acest tutorial, vom acoperi:>> 1. TOC > {:toc} >{: .agendă} # # două căi prin acest tutorialam creat două traiectorii pe care le puteți urmări prin acest tutorial.1. ** Traiectoria 1 * * – Începeți cu SRA NCBI lui și de căutare pentru accesiunile disponibile start-ul (#secventa-read-archive)2. ** Traiectoria 2 * * – bypass SRA NCBI și începe cu Galaxy direct. Start (#înapoi în galaxie)vă recomandăm să începeți cu **traiectoria 2**.# Secvența de citire ArchiveThe (https://www.ncbi.nlm.nih.gov/sra) este arhiva principală a *neasamblate citește* pentru (https://www.ncbi.nlm.nih.gov/). SRA este un loc minunat pentru a obține datele de secvențiere care stau la baza publicațiilor și studiilor.Acest tutorial se referă la modul de a obține date de secvență de la SRA în galaxie, folosind o conexiune directă între cele două.> ### Comentariu Comentariu>> veți auzi, de asemenea, SRA denumit *Arhiva scurtă de citire*, numele său original.>{: .comentariu} # # accesarea SRASRA se poate ajunge fie direct prin intermediul site-ul este, sau prin intermediul panoului de instrumente de pe Galaxy.> ### Comentariu Comentariu>> inițial opțiunea panoului de instrumente pentru accesarea SRA există doar pe (https://usegalaxy.org/). Suportul pentru conectarea directă la SRA va fi inclus în versiunea 20.05 a Galaxy {:.comentariu} > ### hands_on Hands-on: Exploreaza SRA Entrez>> 1. Accesați instanța Galaxy aleasă, cum ar fi una dintre (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) sau oricare alta. (Acest tutorial utilizează usegalaxy.org). > 1. Dacă istoricul dvs. nu este deja gol, atunci începeți un nou istoric (consultați (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) pentru mai multe informații despre istoricul galaxiilor)> 1. ** Faceți clic pe * * ‘Obțineți date’ în partea de sus a panoului de instrumente.> 1. ** Faceți clic pe * * ‘Sra Server `în lista de instrumente afișate sub`Get Data’.> acest lucru vă duce (https://www.ncbi.nlm.nih.gov/sra) – puteți începe, de asemenea, direct de la SRA. O casetă de căutare este afișată în partea de sus a paginii. Încercați să căutați ceva care vă interesează, cum ar fi `dolphin` sau `rinichi` sau `rinichi delfin` și apoi **faceți clic pe** butonul `Căutare`.>> aceasta returnează o listă de *experimente SRA* care se potrivesc cu șirul dvs. de căutare. Experimentele SRA, cunoscute și ca intrări * SRX*, conțin date de secvență dintr-un anumit experiment, precum și o explicație a experimentului în sine și a oricăror alte date conexe. Puteți explora experimentele returnate făcând clic pe numele lor. A se vedea (https://www.ncbi.nlm.nih.gov/Cărți/nbk56913/) în (https://www.ncbi.nlm.nih.gov/Cărți/n/helpsrakb/) pentru mai multe.>> când introduceți text în caseta de căutare SRA, utilizați (https://www.ncbi.nlm.nih.gov/sra/docs/srasearch/). Entrez acceptă atât căutări simple de text, cât și căutări foarte precise care verifică metadate specifice și utilizează expresii logice complexe arbitrar. Entrez vă permite să scalați căutările de la bază la avansat pe măsură ce restrângeți căutările. Sintaxa căutărilor avansate poate părea descurajantă, dar SRA oferă o grafică (https://www.ncbi.nlm.nih.gov/sra/advanced/) pentru a genera sintaxa specifică. Și, după cum vom vedea mai jos, Sra Run Selector oferă o interfață de utilizator și mai prietenoasă pentru îngustarea datelor selectate.>> jucați-vă cu interfața Sra Entrez, inclusiv Generatorul de interogări avansate, pentru a vedea dacă puteți identifica un set de experimente SRA care sunt relevante pentru unul dintre domeniile dvs. de cercetare.{: .hands_on}> # # # hands_on Hands-on: Generați lista experimentelor potrivite folosind Entrez>> acum, că aveți o familiaritate de bază cu Sra Entrez, să găsim secvențele utilizate în acest tutorial.>> 1. Dacă nu sunteți deja acolo, * * navigați * * înapoi la (https://www.ncbi.nlm.nih.gov/sra> 1. ** Clear * * orice text de căutare din caseta de căutare.> 1. ** Tastați * * ‘ sars-COV-2` în caseta de căutare și **faceți clic pe** `Căutare`.> aceasta returnează o listă lungă de experimente SRA care se potrivesc căutării noastre, iar această listă este mult prea lungă pentru a fi utilizată într-un exercițiu tutorial. În acest moment am putea folosi advanced entrez query builder despre care am aflat mai sus.> dar nu o vom face. în schimb, vă permite să trimiteți rezultatele listei *prea lungi pentru un tutorial* pe care le avem la selectorul de rulare SRA și să folosiți interfața sa mai prietenoasă pentru a restrânge rezultatele.>> !(../../ poze / sra_entrez.png) {:.hands_on}> # # # hands_on Hands-on: Treceți de la Entrez la Sra Run Selector>> vizualizați rezultatele ca un tabel interactiv extins folosind RunSelector.>> 1. Faceți clic pe Trimitere rezultate pentru a rula selector, care apare într-o casetă din partea de sus a rezultatelor căutării.>> !(../../ poze / sra_entrez_result.png)>>> # # # sfat ce se întâmplă dacă nu vedeți linkul selectorului de rulare?>>>> este posibil să fi observat acest text mai devreme când explorai căutarea Entrez. Acest text apare doar o parte din timp, când numărul de rezultate ale căutării se încadrează într-o fereastră destul de largă. Nu o veți vedea dacă aveți doar câteva rezultate și nu o veți vedea dacă aveți mai multe rezultate decât poate accepta selectorul de rulare.>>>> *aveți nevoie pentru a obține pentru a rula Selector pentru a trimite rezultatele la Galaxy.* Ce se întâmplă dacă nu aveți suficiente rezultate pentru a declanșa acest link fiind afișate? În acest caz, apelați get to the Run Selector făcând **clic pe** din meniul derulant `Send to` din partea dreaptă sus a panoului de rezultate. Pentru a ajunge la Run Selector, * * selectați * * ‘Run Selector’ și apoi * * faceți clic pe * * butonul ‘Go’.>!(../../ poze / sra_entrez_send_to.png) > {: .sfat}>>> 1. ** Faceți clic pe* * `Trimite rezultate pentru a rula selector ‘ în partea de sus a panoului cu rezultatele căutării. (Dacă nu vedeți acest link, atunci vedeți comentariul direct de mai sus.){: .hands_on} # # Sra Run Selectoram învățat mai devreme cum să restrângem rezultatele căutării folosind sintaxa avansată a lui Entrez. Cu toate acestea, nu am profitat de această putere când eram în Entrez. În schimb, am folosit o căutare simplă și apoi am trimis toate rezultatele către selectorul Run. Nu avem încă lista (scurtă) a rezultatelor pe care dorim să le analizăm. *Ce facem?* Folosim Entrez și selectorul Run modul în care acestea sunt proiectate pentru a fi utilizate: * utilizați interfața Entrez pentru a restrânge rezultatele până la o dimensiune care selectorul Run poate consuma. * Trimiteți aceste rezultate Entrez la selectorul de rulare SRA * utilizați interfața mult mai prietenoasă a selectorului de rulare la 1. Înțelegeți mai ușor datele pe care le avem 1. Restrângeți aceste rezultate folosind aceste cunoștințe.> ### comment Run Selector este atât mai mult cât și mai puțin decât Entrez>> Run Selector poate face cel mai mult, dar nu tot ceea ce poate face sintaxa de căutare Entrez. Run selector utilizează * căutare fațete * tehnologie care este ușor de utilizat, și puternic, dar care are limite inerente. Mai exact, Entrez va funcționa mai bine atunci când căutați atribute care au zeci, sute sau mii de valori diferite. Run Selector va funcționa mai bine căutarea atribute cu mai puțin de 20 de valori diferite. Din fericire, asta descrie majoritatea căutărilor.{: .comentariu}fereastra Run Selector este împărțită în mai multe panouri:* **`listă filtre`**: în colțul din stânga sus. Aici ne vom perfecționa căutarea.* * * ‘Select’**: un rezumat a ceea ce a fost inițial trecut pentru a rula Selector, și cât de mult din care am selectat până în prezent. (Și până acum, nu am selectat niciuna dintre ele.) De asemenea, rețineți butonul tentant, dar încă Gri, `Galaxy`.** * ` S-au găsit X elemente ‘ * * inițial, aceasta este lista de elemente trimise pentru a rula Selector de Entrez. Această listă se va micșora pe măsură ce îi aplicăm filtre.!(../../ poze / sra_run_selector.png) > ### comentariu de ce a crescut numărul de articole găsite*?* >> reamintim că interfața Entrez listează experimentele SRA (intrări SRX). Run Selector liste * ruleaza * – seturi de date secvențiere — și există * una sau mai multe * ruleaza pe experiment. Avem aceleași date ca înainte, acum le vedem doar în detalii mai fine.{: .comentariu} ‘lista filtrelor’ din stânga sus arată coloanele din rezultatele noastre care au fie valori numerice continue, fie 10 sau mai puțin (puteți modifica acest număr) valori distincte în ele. ** Derulați * * în jos prin listă selectați câteva dintre filtre. Când este selectat un filtru, o casetă* valori * apare mai jos, listarea opțiunilor pentru acest filtru și numărul de rulări cu fiecare opțiune. Aceste valori / opțiuni sunt extrase din metadatele setului de date. Încercați * * selectarea * * câteva filtre de sondare interesante și apoi * * selectați * * una sau mai multe opțiuni pentru fiecare filtru. Încercați * * deselectarea * * opțiuni și filtre. Pe măsură ce faceți acest lucru, numărul rezultatelor găsite va scădea sau va crește.> # # # Sfat Sfat: Utilizați filtre pentru a înțelege mai bine datele>> filtrele sunt modul în care restrângeți seturile de date luate în considerare pentru trimiterea către Galaxy, dar sunt, de asemenea, o modalitate excelentă de a înțelege datele dvs.:> în primul rând, selectarea unui filtru este o modalitate ușoară de a vedea intervalul de valori dintr-o coloană. Este posibil să nu puteți (https://www.google.com/search?q=sra+sirs_outcome), dar vă puteți da seama văzând ce valori sunt în el.> în al doilea rând, puteți explora modul în care diferite coloane se raportează între ele. Există o relație între valorile` sirs_outcome `și valorile` disease_stage’?{: .sfat}> ### hands_on Hands-on: restrânge rezultatele folosind Run Selector>> 1. Dacă aveți filtre activate, * * deselectați-le**.> după ce ați făcut acest lucru, nu vor mai apărea casete *valori* sub „lista filtrelor”.> 2. ** Copiați și lipiți * * acest șir de căutare în caseta de căutare` articole găsite’.>> SRR11772204 sau SRR11597145 sau SRR11667145>> această mână-set ales de ruleaza limitează rezultatele noastre la 3 ruleaza din distribuție geografică diferită.{: .hands_on}acest lucru reduce lista `articole găsite ‘ de la zeci de mii de ruleaza la 3 ruleaza (un număr de gestionat pentru un tutorial!). Dar încă nu am terminat cu selectorul de rulare. Rețineți că butonul ‘Galaxy’este încă Gri. Ne-am restrâns opțiunile, dar încă nu am selectat nimic de trimis către Galaxy.Este posibil să selectați fiecare rulare rămasă prin **făcând clic pe** bifa din partea de sus a primei coloane. Puteți deselecta totul prin * * clic * * pe `X`.> ### hands_on Hands-on: selectați rulează și trimite la Galaxy>> 1. Selectați toate ruleaza de * * clic pe * * `X`.> și acum, Butonul „Galaxy” este live.> 1. ** Faceți clic pe butonul ‘Galaxy `din secțiunea` Select’ din partea de sus a paginii.{: .hands_on} # # înapoi în GalaxyWhen facem clic pe ‘Galaxy’ în Run Selector mai multe lucruri se întâmplă. În primul rând, lansează o nouă filă sau fereastră de browser care se deschide în Galaxy. Veți vedea * caseta verde mare * care indică faptul că strângerea de mână dintre SRA și Galaxy a avut succes și veți vedea apoi un nou loc de muncă `SRA` în panoul Istoric. Această casetă poate începe ca gri / în așteptare, indicând faptul că transferul nu a început încă sau poate merge direct la galben / alergare sau la verde / terminat.> # # # hands_on Hands-on: Examinați noul set de date SRA>> 1. Odată ce transferul ‘ SRA ‘ este finalizat, **faceți clic pe** pe pictograma galaxy-eye (eye) a setului de date.>> Aceasta afișează setul de date în panoul central al Galaxy.{: .hands_on}setul de date ‘ SRA ‘ nu este date de secvență, ci mai degrabă *metadate* pe care le vom folosi pentru a obține date de secvență de la SRA. Aceste metadate reflectă informațiile pe care le-am văzut în secțiunea `articole găsite` a selectorului Run. Metadatele nu sunt datele finale pe care le căutăm de la SRA, dar având toate metadatele este adesea utilă în etapele ulterioare de analiză.Să folosim acum metadatele pentru a prelua datele secvenței de la SRA. SRA oferă instrumente pentru extragerea tot felul de informații, inclusiv datele de secvență în sine. Instrumentul Galaxy ‘descărcare și extragere mai rapidă citește în FASTQ’ se bazează pe utilitarul SRA (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump) și face exact asta.– >
- Găsiți datele necesare în SRA
- hands_on Hands-on: descrierea sarcinii
- Comentariu Comentariu
- proces și filtru SraRunInfo.fișier csv în Galaxy
- hands_on Hands-on: încărcați SraRunInfo.fișier csv în Galaxy
- comentariu Feriți-vă de tăieturi
- hands_on Hands-on: crearea unui subset de date
- Sfat Sfat: găsirea instrumente
- descărcați secvențierea datelor cu descărcare și extragere mai rapidă citește în FASTQ
- hands_on Hands-on: descrierea sarcinii
- acum ce?
- analiza variației datelor de secvențiere SARS-Cov-2
- comentariu usegalaxy.* Proiect de analiză COVID-19
- obțineți datele genomului de referință
- hands_on Hands-on: obțineți datele genomului de referință
- sfat: importul prin link-uri
- tunderea adaptorului cu fastp
- hands_on Hands-on: descriere sarcină
- alinierea cu harta cu BWA-mem
- hands_on Hands-on: Aliniați secvențierea citește la genomul de referință
- eliminați duplicatele cu MarkDuplicates
- hands_on Hands-on: eliminați duplicatele PCR
- generați statistici de aliniere cu samtools stats
- hands_on Hands-on: Generați statistici de aliniere
- Realign citește cu lofreq viterbi
- hands_on Hands-on: Realign citește în jurul indels
- adăugați calități indel cu lofreq introduceți calități indel
- hands_on Hands-on: Adăugați calități indel
- apel variante folosind variante de apel lofreq
- hands_on Hands-on: variante de apel
- Adnotați efectele variantei cu SnpEff eff:
- hands_on Hands – on: Adnotați efectele variantei
- creați tabelul de variante folosind câmpurile de extragere SnpSift
- hands_on Hands-on: creați tabelul de variante
- rezumați datele cu MultiQC
- hands_on Hands-on: rezumați datele
- concluzie
- keypoints puncte cheie
- Întrebări frecvente
- literatură utilă
- Feedback
- citând acest Tutorial
- details BibTeX
Găsiți datele necesare în SRA
Mai întâi trebuie să găsim un set de date bun cu care să ne jucăm. Arhiva de citire a secvenței (Sra) este arhiva principală a citirilor neasamblate operate de Institutele Naționale de sănătate din SUA (NIH). SRA este un loc minunat pentru a obține datele de secvențiere care stau la baza publicațiilor și studiilor. Să facem asta:
hands_on Hands-on: descrierea sarcinii
- accesați pagina SRA a NCBI îndreptând browserul către https://www.ncbi.nlm.nih.gov/sra
- în caseta de căutare introduceți
SARS-CoV-2 Patient Sequencing From Partners / MGH(alternativ, pur și simplu faceți clic pe acest link)
- pagina web va afișa un număr mare de seturi de date SRA (la momentul scrierii erau 2.223). Acestea sunt date dintr-un studiu care descrie analiza SARS-CoV-2 în zona Boston.
- descărcați metadatele care descriu aceste seturi de date prin:
- făcând clic pe Trimite la: dropdown
- selectând
File- Schimbarea formatului în
RunInfo- făcând clic pe Creare fișieraici este cum ar trebui să arate:
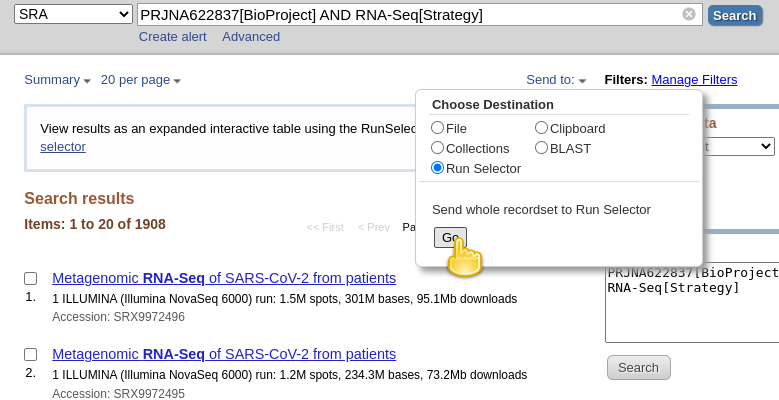
- aceasta ar crea un fișier
SraRunInfo.csvîn folderulDownloadsdestul de mare.
acum că am descărcat acest fișier, putem merge la o instanță Galaxy și să începem procesarea acestuia.
Comentariu Comentariu
rețineți că fișierul pe care tocmai l-am descărcat nu este secvențierea datelor în sine. Mai degrabă, este metadate care descriu proprietățile de secvențiere citește. Vom filtra această listă până la doar câteva aderări care vor fi utilizate în restul acestui tutorial.
proces și filtru SraRunInfo.fișier csv în Galaxy
hands_on Hands-on: încărcați SraRunInfo.fișier csv în Galaxy
- du-te la instanță Galaxy de alegere, cum ar fi unul dintre usegalaxy.org, usegalaxy.eu, usegalaxy.org.au sau oricare alta. (Acest tutorial utilizează usegalaxy.org).
- Faceți clic pe butonul Încărcare date:
- în caseta de dialog care va apărea Faceți clic pe butonul „Alegeți fișierele locale”:

- găsiți și selectați
SraRunInfo.csvfișier de pe computer- Faceți clic pe butonul Start
- închideți butonul
- acum puteți privi conținutul acestui fișier făcând clic pe pictograma Galaxy-eye (eye). Veți vedea că acest fișier conține o mulțime de informații despre aderările individuale SRA. În acest studiu, fiecare aderare corespunde unui pacient individual ale cărui probe au fost secvențiate.
Galaxy poate procesa toate cele 2.000 de seturi de date, dar pentru a face acest tutorial suportabil trebuie să selectăm un subset mai mic. În special experiența noastră anterioară cu aceste date arată două seturi de date interesante SRR11954102 și SRR12733957. Deci, hai să le scoatem.
comentariu Feriți-vă de tăieturi
secțiunea Hands-on de mai jos folosește instrumentul de tăiere. Există două instrumente tăiate în Galaxy din motive istorice. Acest exemplu utilizează instrumentul cu numele complet tăiați coloanele dintr-un tabel (tăiat). Cu toate acestea, aceeași logică se aplică și celuilalt instrument. Pur și simplu are o interfață ușor diferită.
hands_on Hands-on: crearea unui subset de date
- Find tool „Select lines that match an expression” instrument în secțiunea filtru și sortare a panoului de instrumente.
Sfat Sfat: găsirea instrumente
Galaxy poate avea o cantitate copleșitoare de instrumente instalate. Pentru a găsi un anumit instrument, tastați numele instrumentului în caseta de căutare a panoului de instrumente pentru a găsi instrumentul.
- asigurați-vă că
SraRunInfo.csvsetul de date pe care tocmai l-am încărcat este listat în câmpul param-file „Select lines from” Al formularului de instrument.- în câmpul „Model” introduceți următoarea expresie
SRR12733957|SRR11954102. Acestea sunt două aderări pe care dorim să le găsim separate de simbolul conductei||înseamnăor: găsiți linii care conținSRR12733957sauSRR11954102.- Faceți clic pe
Executebuton.- acest lucru va genera un fișier care conține două linii (bine … o linie este, de asemenea, utilizat ca antet, astfel încât acesta va apărea fișierul are trei linii. Este OK.)
- tăiați prima coloană din fișier folosind instrumentul instrument „Cut”, pe care îl veți găsi în secțiunea de manipulare a textului din panoul de instrumente.
- asigurați-vă că setul de date produs de pasul anterior este selectat în câmpul „Fișier de tăiat” al formularului de instrument.
- schimbați „delimitat de” la
Comma- în „lista câmpurilor” selectați
Column: 1.- Hit
Executeacest lucru va produce un fișier text cu doar două linii:SRR12733957SRR11954102
acum, că avem identificatori de seturi de date pe care vrei avem nevoie pentru a descărca datele reale secvențiere.
descărcați secvențierea datelor cu descărcare și extragere mai rapidă citește în FASTQ
hands_on Hands-on: descrierea sarcinii
- descărcare și extragere mai rapidă citește în instrumentul FASTQ cu următorii parametri:
- „selectați tipul de intrare”:
List of SRA accession, one per line
- parametrul param-file „lista de aderare sra” ar trebui să indice ieșirea instrumentului „Cut” din pasul anterior.
- Faceți clic pe butonul
Execute. Aceasta va rula instrumentul, care preia seturile de date de citire a secvenței pentru rulările care au fost listate în setul de dateSRA. Poate dura ceva timp. Deci, acest lucru poate fi un moment bun pentru a obține cafea.- Mai multe intrări sunt create în panoul Istoric atunci când trimiteți acest job:
Pair-end data (fasterq-dump): Conține seturi de date cu capăt asociat (dacă sunt disponibile)Single-end data (fasterq-dump)conține seturi de date cu capăt unic (dacă sunt disponibile)Other data (fasterq-dump)conține seturi de date nepereche (dacă sunt disponibile)fasterq-dump logconține informații despre executarea instrumentului
primele trei elemente sunt de fapt colecții de seturi de date. Colecțiile din Galaxy sunt grupări logice de seturi de date care reflectă relațiile semantice dintre ele în experiment / analiză. În acest caz, instrumentul creează o colecție separată pentru fiecare pereche-end citește, singur citește, și altele.Consultați tutorialele colecțiilor pentru mai multe.
explorați colecțiile făcând mai întâi clic pe numele colecției din panoul Istoric. Acest lucru vă duce în interiorul colecției și vă arată seturile de date din ea. Apoi puteți naviga înapoi la nivelul exterior al istoriei.
odată cefasterq termină transferul de date (toate casetele sunt verzi / terminate), suntem gata să le analizăm.
acum ce?
acum Puteți analiza datele preluate folosind orice instrumente de analiză a secvențelor și fluxuri de lucru din Galaxy. SRA deține date de suport pentru fiecare tip imaginabil de experiment *-seq.
dacă ați rulat acest tutorial, dar ați recuperat seturi de date care v-au interesat, consultați restul bibliotecii GTN pentru idei despre cum să analizați în Galaxy.
cu toate acestea, dacă ați preluat seturile de date utilizate în exemplele acestui tutorial de mai sus, atunci sunteți gata să rulați analiza variantei SARS-CoV-2 de mai jos.
analiza variației datelor de secvențiere SARS-Cov-2
În această parte a tutorialului vom efectua apelarea variantelor și analiza de bază a seturilor de date descărcate mai sus. Vom începe prin descărcarea secvenței de referință Wuhan-Hu-1 SARS-CoV-2, Apoi vom rula tunderea adaptorului, alinierea și apelarea variantelor și, în final, vom analiza distribuția geografică a unora dintre variantele găsite.
comentariu usegalaxy.* Proiect de analiză COVID-19
acest tutorial folosește un subset de date și rulează prin analiza variației covid19.galaxyproject.org.Datele pentru covid19.galaxyproject.org este actualizat continuu pe măsură ce noile seturi de date sunt făcute publice.
obțineți datele genomului de referință
datele genomului de referință pentru astăzi sunt pentru SARS-CoV-2, „sindrom respirator acut sever coronavirus 2 izolat Wuhan-Hu-1, genom complet”, având ID-ul de aderare al NC_045512.2.
aceste date sunt disponibile de la Zenodo folosind următorul link.
hands_on Hands-on: obțineți datele genomului de referință
importați următorul fișier în istoricul dvs:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gzsfat: importul prin link-uri
- copiați locația link-ul
- deschideți Galaxy Upload Manager (galaxy-upload pe partea din dreapta sus a panoului de instrumente)
- selectați Paste/Fetch date
- lipiți linkul în câmpul text
- apăsați Start
- închideți ferestreaîn mod implicit, Galaxy folosește adresa URL ca nume, redenumiți fișierele cu un nume mai util.
tunderea adaptorului cu fastp
eliminarea adaptoarelor de secvențiere îmbunătățește alinierile și apelarea variantelor. instrumentul fastp poate detecta automat adaptoarele de secvențiere utilizate pe scară largă.
hands_on Hands-on: descriere sarcină
- instrument fastp cu următorii parametri:
- „single-end sau asociat citește”:
Paired Collection
- param-file ” selectați colecție asociat(e)”:
list_paired(ieșire de descărcare și extragere mai rapidă citește în instrumentul FASTQ)- în „opțiuni de ieșire”:
- „raport de ieșire JSON”:
Yes
alinierea cu harta cu BWA-mem
instrumentul BWA-mem este un Aligner de secvență utilizat pe scară largă pentru seturi de date de secvențiere citite scurt, cum ar fi cele pe care le analizăm în acest tutorial.
hands_on Hands-on: Aliniați secvențierea citește la genomul de referință
- harta cu instrumentul BWA-mem cu următorii parametri:
- „veți selecta un genom de referință din istoricul dvs. sau veți utiliza un index încorporat?”:
Use a genome from history and build index
- param-file „folosiți următorul set de date ca secvență de referință”:
output(set de date de intrare)- „citire simplă sau pereche”:
Paired Collection
- param-fișier „selectați o colecție asociat”:
output_paired_coll(ieșire de instrument fastp)- „set citit grupuri de informații?”:
Do not set- „selectați modul de analiză”:
1.Simple Illumina mode
eliminați duplicatele cu MarkDuplicates
instrumentul MarkDuplicates elimină secvențele duplicate originare din artefacte de pregătire a bibliotecii și artefacte de secvențiere. Este important să eliminați aceste secvențe artefactuale pentru a evita suprareprezentarea artificială a unei singure molecule.
hands_on Hands-on: eliminați duplicatele PCR
- instrumentul MarkDuplicates cu următorii parametri:
- param-file „Select SAM/BAM dataset or dataset collection”:
bam_output(ieșirea hărții cu instrumentul BWA-MEM)- „dacă este adevărat nu scrieți duplicate în fișierul de ieșire în loc să le scrieți cu set de steaguri adecvate”:
Yes
generați statistici de aliniere cu samtools stats
după pasul de marcare duplicat de mai sus putem genera statistici despre alinierea pe care am generat-o.
hands_on Hands-on: Generați statistici de aliniere
- Samtools stats tool cu următorii parametri:
- param-file „BAM file”:
outFile(output of MarkDuplicates tool)- „Set coverage distribution”:
No- „Output”:
One single summary file- „filtru de steaguri Sam”:
Do not filter- „utilizați o secvență de referință”:
No- „filtru de regiuni”:
No
Realign citește cu lofreq viterbi
Realign citește instrument corectează nealinieri în jurul insertii si stergeri. Acest lucru este necesar pentru a detecta cu exactitate variantele.
hands_on Hands-on: Realign citește în jurul indels
- Realign citește cu instrumentul lofreq cu următorii parametri:
- param-file „citește la realign”:
outFile(ieșirea instrumentului MarkDuplicates)- „alegeți sursa pentru genomul de referință”:
History
- param-fișier” referință”:
output(set de date de intrare)- în” opțiuni avansate”:
- ” cum să se ocupe de calitățile de bază ale 2?”:
Keep unchanged
adăugați calități indel cu lofreq introduceți calități indel
acest pas adaugă calități indel în fișierul nostru de aliniere. Acest lucru este necesar pentru a apela variante folosind variante de apel cu instrumentul lofreq
hands_on Hands-on: Adăugați calități indel
- introduceți calități indel cu instrumentul lofreq cu următorii parametri:
- param-file „Citește”:
realigned(ieșirea instrumentului Realign citește)- „abordare de calcul Indel”:
Dindel
- „Alegeți sursa pentru genomul de referință”:
History
- param-fișier „referință”:
output(set de date de intrare)
apel variante folosind variante de apel lofreq
suntem acum gata pentru a apela variante.
hands_on Hands-on: variante de apel
- variante de apel cu instrumentul lofreq cu următorii parametri:
- param-file „Input citește în format BAM”:
output(ieșirea instrumentului Insert Indel calities)- „Alegeți sursa pentru genomul de referință”:
History
- Param-fișier „referință”:
output(set de date de intrare)- „variante de apel peste”:
Whole reference- „tipuri de variante pentru a apela”:
SNVs and indels- „variant calling parameters”:
Configure settings
- În „Coverage”:
- „minimal coverage”:
50- În „base-calling”:
- „minim baseq”:
30- „minim baseq pentru baze alternative”:
30- în „mapping qualityy
20- „parametrii filtrului variant”:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
rezultatul acestui pas este o colecție de fișiere VCF care pot fi vizualizate într-un browser genom.
Adnotați efectele variantei cu SnpEff eff:
vom adnota acum variantele pe care le-am numit în pasul anterior cu efectul pe care îl au asupra genomului SARS-CoV-2.
hands_on Hands – on: Adnotați efectele variantei
- SnpEff eff: instrument cu următorii parametri:
- param-file ” modificări de secvență (SNPs, MNPs, InDels)”:
variants(instrument de ieșire a variantelor de apel)- „format de ieșire”:
VCF (only if input is VCF)- „creați un raport CSV, util pentru analiza din aval (-csvStats)”:
Yes- „Opțiuni de adnotare”: `
- „filter output”: `
- „filter out specific effects”:
No
ieșirea acestui pas este un fișier VCF cu efecte variante adăugate.
creați tabelul de variante folosind câmpurile de extragere SnpSift
acum vom selecta diferite efecte din VCF și vom crea un fișier tabular mai ușor de înțeles pentru oameni.
hands_on Hands-on: creați tabelul de variante
- Snpsift Extract Fields tool cu următorii parametri:
- param-file „fișier de intrare variantă în format VCF”:
snpeff_output(ieșire de SnpEff eff: instrument)- „câmpuri de extras”:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- „separator de câmpuri multiple”:
,- „text câmp gol”:
.
putem inspecta fișierele de ieșire și a vedea verifica dacă variantele din acest fișier sunt, de asemenea, descrise într-un notebook observabil care arată distribuția geografică a secvențelor variantei SARS-COV-2
variantele interesante includ varianta C la t la poziția 14408 (14408c/T) în srr11772204, 28144t/C în srr11597145 și 25563g/T în srr11667145.
rezumați datele cu MultiQC
vom rezuma acum analiza noastră cu MultiQC, care generează un raport frumos pentru datele noastre.
hands_on Hands-on: rezumați datele
- instrument MultiQC cu următorii parametri:
- În” rezultate”:
- param-repeat” Insert Results „
- ” ce instrument a fost folosit generați jurnale?”:
fastp
- param-fișier „ieșire de fastp”:
report_json(ieșire de instrument fastp)- param-repeat” Inserare rezultate „
- ” ce instrument a fost folosit genera jurnale?”:
Samtools
- În” Samtools output”:
- param-repeat” Insert samtools output „
- ” tip de samtools output?”:
stats
- param-file „Samtools statistici ieșire”:
output(ieșire de Samtools statistici instrument)param-repeat” Inserare rezultate „
- ” ce instrument a fost folosit genera jurnale?”:
Picard
- În” ieșire Picard”:
- param-repeat” introduceți ieșire Picard „
- ” Tipul de ieșire Picard?”:
Markdups- param-fișier”ieșire Picard”:
metrics_file(ieșirea instrumentului MarkDuplicates)param-repeat” Insert Results „
- ” ce instrument a fost folosit generați jurnale?”:
SnpEff
- param-fișier” ieșire de SnpEff”:
csvFile(ieșire de SnpEff eff: instrument)
concluzie
felicitări, acum știți cum să importați date de secvență din SRA și cum să rulați un exemplu de analiză pe aceste seturi de date.
keypoints puncte cheie
datele secvenței din SRA pot fi importate direct în Galaxy
Întrebări frecvente
aveți întrebări despre acest tutorial? Consultați pagina de întrebări frecvente pentru subiectul de analiză a variantelor pentru a vedea dacă întrebarea dvs. este listată acolo. Dacă nu, vă rugăm să puneți întrebarea dvs. pe canalul GTN Gitter sau pe forumul de ajutor Galaxy
literatură utilă
informații suplimentare, inclusiv link-uri către documentație și publicații originale, cu privire la instrumentele, tehnicile de analiză și interpretarea rezultatelor descrise în acest tutorial pot fi găsite aici.
Feedback
ați folosit acest material ca instructor? Simțiți-vă liber să ne oferiți feedback despre cum a mers.
citând acest Tutorial
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 din arhiva de citire a secvenței NCBI (SRA) la Galaxy: SARS-COV-2 variant analysis (Galaxy Training Materials). / instruire-material/subiecte/variantă-analiză/tutoriale/sars-cov-2 / tutorial.html Online; accesat astăzi
- Batut și colab., 2018 instruire de analiză a datelor bazată pe comunitate pentru sistemele celulare de Biologie 10.1016/j.cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}