syftet med denna handledning är att introducera dig till behandlingen av nästa generations sekvenseringsdata i Galaxy. Denna handledning använder en COVID-19-variant som ringer från Illumina data, men det handlar inte om variantsamtal i sig.
vid slutförandet av denna handledning kommer du att veta:
- hur man hittar data i SRA och överför denna information till Galaxy
- hur man utför grundläggande ngs-databehandling i Galaxy inklusive:
- kvalitetskontroll (QC) av Illumina data
- kartläggning
- borttagning av dubbletter
- Variant ringer med
lofreq - Variant annotation
- använda datamängder samlingar
- importera data till Jupyter
### Agenda>> I denna handledning kommer vi att täcka:>> 1. TOC> {: toc}> {: .agenda} # # två vägar genom denna handledningvi skapade twoi banor som du kan följa genom denna handledning.1. ** Trajectory 1 * * – Börja med NCBI SRA och söka efter tillgängliga anslutningar bisexuell Start (#the-sekvens-läs-arkiv)2. ** Bana 2 * * – bypass NCBI SRA och börja med Galaxy direkt. Start (#back-in-galaxy)vi rekommenderar att du börjar med **Trajectory 2**.# Sekvensen läsa Arkivethe (https://www.ncbi.nlm.nih.gov/sra) är den primära arkiv *omonterade läser* för (https://www.ncbi.nlm.nih.gov/). SRA är ett bra ställe att få sekvenseringsdata som ligger till grund för publikationer och studier.Denna handledning täcker hur man får sekvensdata från SRA till Galaxy med en direkt koppling mellan de två.> ### comment Comment>> du kommer också att höra SRA kallas *Kortläst Arkiv*, dess ursprungliga namn.>{: .kommentar} # # åtkomst till SRASRA kan nås antingen direkt via sin webbplats eller via verktygspanelen på Galaxy.> ### comment Comment>> ursprungligen finns verktygspanelalternativet för åtkomst till SRA endast på (https://usegalaxy.org/). Stöd för direktanslutning till SRA kommer att ingå i 20.05-utgåvan av Galaxy{: .kommentar}> # # # hands_on Hands-on: utforska SRA entrez>> 1. Gå till din Galaxy-instans som en av (https://usegalaxy.org/https://usegalaxy.euhttps://usegalaxy.org.au) eller någon annan. (Denna handledning använder usegalaxy.org).> 1. Om historiken inte redan är tom, än starta en ny historia (se (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/history/tutorial.html) för mer information om Galaxy historier)> 1. ** Klicka på * * ’Hämta Data’ högst upp på verktygspanelen.> 1. ** Klicka på * * ’Sra Server’ i listan över verktyg som visas under ’hämta Data’.> detta tar dig (https://www.ncbi.nlm.nih.gov/sra) – du kan också starta direkt från SRA. En sökruta visas högst upp på sidan. Försök att söka efter något du är intresserad av, till exempel `dolphin` eller `kidney` eller `dolphin kidney` och sedan **klicka** på `Sök` – knappen.>> detta returnerar en lista med *Sra-experiment* som matchar din söksträng. Sra-experiment, även kända som * SRX-poster*, innehåller sekvensdata från ett visst experiment, samt en förklaring av själva experimentet och andra relaterade data. Du kan utforska de returnerade experimenten genom att klicka på deras namn. Se (https://www.ncbi.nlm.nih.gov/böcker/NBK56913/) i (https://www.ncbi.nlm.nih.gov/böcker/n/helpsrakb/) för mer.>> när du skriver in text i SRA-sökrutan använder du (https://www.ncbi.nlm.nih.gov/sra/docs/srasearch/). Entrez stöder både enkla textsökningar och mycket exakta sökningar som kontrollerar specifika metadata och använder godtyckligt komplexa logiska uttryck. Med Entrez kan du skala upp dina sökningar från grundläggande till avancerade när du begränsar dina sökningar. Syntaxen för avancerade sökningar kan verka skrämmande, men SRA ger en grafisk (https://www.ncbi.nlm.nih.gov/sra/advanced/) för att generera den specifika syntaxen. Och som vi ska se nedan ger Sra Run Selector ett ännu vänligare användargränssnitt för att begränsa våra valda data.>> lek med sra Entrez-gränssnittet, inklusive advanced query builder, för att se om du kan identifiera en uppsättning SRA-experiment som är relevanta för ett av dina forskningsområden.{: .hands_on}> # # # hands_on Hands-on: Generera lista över matchande experiment med Entrez>> nu när du har en grundläggande kännedom om SRA Entrez, låt oss hitta sekvenserna som används i denna handledning.>> 1. Om du inte redan är där, **navigera * * tillbaka till (https://www.ncbi.nlm.nih.gov/sra> 1. ** Rensa * * någon söktext från sökrutan.> 1. ** Skriv* * `sars-cov-2 `i sökrutan och **klicka**`Sök’.> detta returnerar en lång lista över sra-experiment som matchar vår sökning, och den listan är alldeles för lång för att användas i en handledning. Vid denna tidpunkt kunde vi använda advanced entrez query builder som vi lärde oss om ovan.> men vi kommer inte. låt oss istället skicka *för länge för en handledning* lista resultat vi har till Sra Run-Väljaren och använd dess vänligare gränssnitt för att begränsa våra resultat.>> !(../../ bilder / sra_entrez.png) {:.hands_on}> # # # hands_on Hands-on: Gå från Entrez till Sra Run Selector>> visa resultat som en utökad interaktiv tabell med RunSelector.>> 1. Klicka på Skicka resultat för att köra väljare, som visas i en ruta högst upp i sökresultaten.>> !(../../ bilder / sra_entrez_resultat.png)>>> ### Tips Vad händer om du inte ser länken Kör väljare?>>>> du kanske har märkt den här texten tidigare när du utforskade Entrez-sökning. Den här texten visas bara en del av tiden, när antalet sökresultat faller inom ett ganska brett fönster. Du kommer inte se det om du bara har några resultat, och du kommer inte se det om du har fler resultat än Körväljaren kan acceptera.>>>> *du måste köra väljaren för att skicka dina resultat till Galaxy.* Vad händer om du inte har tillräckligt med resultat för att utlösa denna länk visas? I så fall ringer du till Körväljaren genom att **klicka på** på rullgardinsmenyn `skicka till` längst upp till höger på resultatpanelen. För att komma till Run Selector, * * välj * * ’Run Selector’ och sedan * * klicka * * på` Go ’ – knappen.> !(../../ bilder / sra_entrez_send_to.png)> {: .tips}>>> 1. ** Klicka på * * ’Skicka resultat för att köra väljare’ högst upp på sökresultatpanelen. (Om du inte ser den här länken, se kommentaren direkt ovan.){: .hands_on} # # Sra Run Selectorvi lärde oss tidigare hur vi kan begränsa våra sökresultat genom att använda Entrez avancerade syntax. Vi utnyttjade dock inte den kraften när vi var i Entrez. Istället använde vi en enkel sökning och skickade sedan alla resultat till Körväljaren. Vi har ännu inte den (korta) listan över resultat vi vill köra analys på. * Vad gör vi?* Vi använder Entrez och Körväljaren hur de är utformade för att användas: * Använd entrez-gränssnittet för att begränsa dina resultat till en storlek som Körväljaren kan konsumera. * Skicka dessa Entrez resultat till Sra Run Selector * använd kör väljarens mycket vänligare gränssnitt till 1. Lättare att förstå de data vi har 1. Begränsa dessa resultat med hjälp av den kunskapen.> ### comment Run Selector är både mer och mindre än entrez>> Run Selector kan göra det mesta, men inte allt vad Entrez söksyntax kan göra. Run selector använder * facetterad sökning * teknik som är lätt att använda, och kraftfull, men som har inneboende gränser. Specifikt fungerar Entrez bättre när du söker på attribut som har tiotals, hundratals eller tusentals olika värden. Körväljaren fungerar bättre att söka attribut med färre än 20 olika värden. Lyckligtvis beskriver det de flesta sökningar.{: .kommentar}fönstret Körväljare är uppdelat i flera paneler:* **`filterlista`**: i det övre vänstra hörnet. Det är här vi kommer att förfina vår sökning.* * * ’Select’**: en sammanfattning av vad som ursprungligen skickades till Run Selector, och hur mycket av det vi har valt hittills. (Och hittills har vi inte valt något av det.) Notera också den tantalizing, men fortfarande gråtonad, `Galaxy` – knappen.* * * ’Found x Items’ * * ursprungligen är detta listan över objekt som skickas till Run Selector från Entrez. Denna lista kommer att krympa när vi tillämpar filter på den.!(../../ bilder / sra_run_selector.png)> ### kommentar varför gick antalet hittade objekt*upp ?*>> kom ihåg att Entrez-gränssnittet listar SRA-experiment (SRX-poster). Kör Väljarlistor * körningar — – sekvenseringsdataset-och det finns *en eller flera* körningar per experiment. Vi har samma data som tidigare, vi ser det nu bara i finare detalj.{: .kommentar}listan ’Filter’ uppe till vänster visar kolumner i våra resultat som har antingen kontinuerliga numeriska värden eller 10 eller mindre (Du kan ändra detta nummer) distinkta värden i dem. ** Bläddra * * ner genom listan Välj några av filtren. När ett filter är markerat visas en ruta *värden* nedan, med alternativ för det här filtret och antalet körningar med varje alternativ. Dessa värden / alternativ hämtas från datauppsättningsmetadata. Försök * * välja * * några intressanta ljudfilter och sedan** välj * * ett eller flera alternativ för varje filter. Försök * * avmarkera * * alternativ och filter. När du gör detta kommer antalet hittade resultat att minska eller öka.> ### tips tips: Använd filter för att bättre förstå data>> Filter är hur du begränsar datamängderna som övervägs för att skicka till Galaxy, men de är också ett utmärkt sätt att förstå dina data:> för det första är att välja ett filter ett enkelt sätt att se värdena i en kolumn. Du kanske inte kan (https://www.google.com/search?q=sra+sirs_outcome), men du kan eventuellt räkna ut det genom att se vilka värden som finns i den.> för det andra kan du utforska hur olika kolumner relaterar till varandra. Finns det ett samband mellan `sirs_outcome` – värden och `disease_stage` – värden?{: .tips}> ### hands_on Hands-on: begränsa dina resultat med Run Selector>> 1. Om du har några filter påslagna, **avmarkera * * dem.> när du har gjort det kommer det inte att finnas några *värden* – rutor under ”filterlistan”.> 2. ** Kopiera och klistra in * * denna söksträng i sökrutan` hittade objekt’.>> SRR11772204 eller SRR11597145 eller SRR11667145>> denna hand-plockad uppsättning körningar begränsar våra resultat till 3 körningar från olika geografiska fördelningar.{: .hands_on}detta minskar din` hittade objekt ’ lista från tiotusentals körningar till 3 körningar (ett hanterbart antal för en handledning!). Men vi är inte riktigt färdiga med Run Selector än. Observera att` Galaxy ’ – knappen fortfarande är gråtonad. Vi har minskat våra alternativ, men vi har faktiskt inte valt något att skicka till Galaxy än.Det är möjligt att välja varje återstående körning genom att **klicka på** bocken högst upp i den första kolumnen. Du kan avmarkera allt genom att** klicka på * * `X’.> ### hands_on Hands-on: välj körningar och skicka till Galaxy>> 1. Välj alla körningar genom att** klicka på * * `X’.> och nu är ”Galaxy” – knappen live.> 1. ** Klicka på * * ’Galaxy’ – knappen i avsnittet ’Välj’ högst upp på sidan.{: .hands_on} # # tillbaka i Galaxennär vi klickar på ’Galaxy’ I Run Selector händer flera saker. Först lanserar det en ny webbläsarflik eller ett fönster som öppnas i Galaxy. Du kommer att se den *stora gröna rutan * som indikerar att handskakningen mellan Sra och Galaxy var framgångsrik och du kommer då att se ett nytt `SRA` – jobb i din historikpanel. Den här rutan kan börja som grå / väntande, vilket indikerar att överföringen ännu inte har startat, eller det kan gå direkt till gul / kör eller till grön / klar.> ### hands_on Hands-on: Undersök det nya SRA-datasetet>> 1. När ’ sra ’- överföringen är klar, **klicka* * på dataset ’ s galaxy-eye (eye) – ikon.>> detta visar datauppsättningen i galaxens mittpanel.{: .hands_on} ’ sra ’ dataset är inte sekvensdata, utan snarare * metadata * som vi kommer att använda för att få sekvensdata från SRA. Denna metadata speglar den information vi såg i Körväljarens ’hittade objekt’ avsnitt. Metadata är inte slutdata som vi söker från SRA, men att ha all den metadata är ofta användbar i efterföljande analyssteg.Låt oss nu använda metadata för att hämta sekvensdata från SRA. SRA tillhandahåller verktyg för att extrahera all slags information, inklusive själva sekvensdata. Galaxy-verktyget ”snabbare nedladdning och Extract läser i FASTQ”är baserat på verktyget SRA (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump) och gör just det.– >
- hitta nödvändiga data i SRA
- hands_on Hands-on: Uppgiftsbeskrivning
- kommentar kommentar
- bearbeta och filtrera SraRunInfo.csv-fil i Galaxy
- hands_on Hands-on: Ladda upp SraRunInfo.CSV-fil i Galaxy
- kommentar akta dig för skärningar
- hands_on Hands-on: skapa en delmängd av data
- Tips Tips: Hitta verktyg
- hämta sekvenseringsdata med snabbare nedladdning och extrahera läser i FASTQ
- hands_on Hands-on: Uppgiftsbeskrivning
- nu vad?
- Variationsanalys av SARS-Cov-2 sekvenseringsdata
- kommentera usegalaxy.* COVID-19 analysprojekt
- få referensgenomdata
- hands_on Hands-on: hämta referensgenomdata
- tips: importera via länkar
- Adapter trimning med fastp
- hands_on Hands-on: Uppgiftsbeskrivning
- justering med karta med BWA-mem
- hands_on Hands-on: Justera sekvensering läser till referensgenom
- ta bort dubbletter med Markduplikat
- hands_on Hands-on: ta bort PCR dubbletter
- generera justeringsstatistik med samtools stats
- hands_on Hands-on: Generera justeringsstatistik
- Realign läser med lofreq viterbi
- hands_on Hands-on: Realign läser runt indels
- Lägg till indel-kvaliteter med lofreq infoga indel-egenskaper
- hands_on Hands-on: Lägg indel kvaliteter
- anropsvarianter med lofreq Call variants
- hands_on Hands-on: Samtalsvarianter
- kommentera varianteffekter med SnpEff eff:
- hands_on Hands-on: kommentera varianteffekter
- Skapa tabell med varianter med SnpSift-Extraktfält
- hands_on Hands-on: Skapa tabell med varianter
- sammanfatta data med MultiQC
- hands_on Hands-on: sammanfatta data
- slutsats
- keypoints nyckelpunkter
- Vanliga frågor
- användbar litteratur
- Feedback
- citerar denna handledning
- details BibTeX
hitta nödvändiga data i SRA
först måste vi hitta en bra dataset att spela med. Sequence Read Archive (SRA) är det primära arkivet för omonterade läsningar som drivs av US National Institutes of Health (NIH). SRA är ett bra ställe att få sekvenseringsdata som ligger till grund för publikationer och studier. Låt oss göra det:
hands_on Hands-on: Uppgiftsbeskrivning
- gå till NCBIS sra-sida genom att peka din webbläsare på https://www.ncbi.nlm.nih.gov/sra
- i sökrutan ange
SARS-CoV-2 Patient Sequencing From Partners / MGH(alternativt klickar du bara på den här länken)
- webbsidan visar ett stort antal sra-dataset (i skrivande stund var det 2,223). Detta är data från en studie som beskriver analys av SARS-CoV-2 i Boston-området.
- ladda ner metadata som beskriver dessa datamängder genom att:
- Klicka på Skicka till: dropdown
- välja
File- ändra Format till
RunInfo- Klicka på Skapa filhär är hur det ska se ut:
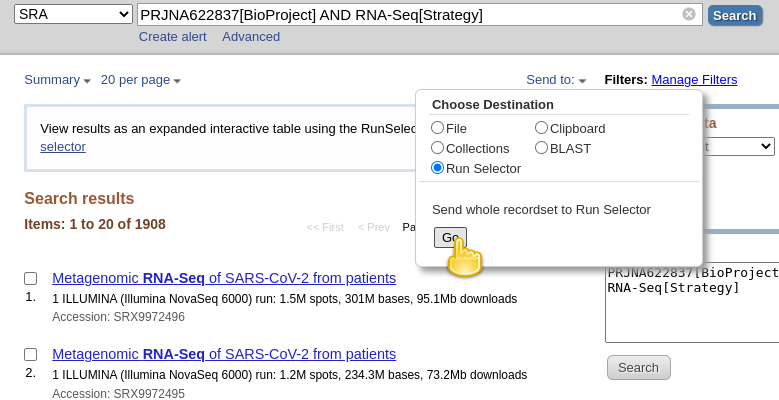
- detta skulle skapa en ganska stor
SraRunInfo.csvfil i dinDownloadsmapp.
Nu när vi har laddat ner den här filen kan vi gå till en Galaxy-instans och börja bearbeta den.
kommentar kommentar
Observera att filen vi just laddade ner inte är sekvenseringsdata i sig. Snarare är det metadata som beskriver egenskaper för sekvenseringsläsningar. Vi kommer att filtrera denna lista ner till bara några anslutningar som kommer att användas i resten av denna handledning.
bearbeta och filtrera SraRunInfo.csv-fil i Galaxy
hands_on Hands-on: Ladda upp SraRunInfo.CSV-fil i Galaxy
- gå till din Galaxy-instans av val som en av usegalaxy.org, usegalaxy.eu, usegalaxy.org.au eller någon annan. (Denna handledning använder usegalaxy.org).
- Klicka på knappen Ladda upp Data:
- i dialogrutan som visas klickar du på knappen ”Välj lokala filer”:

- hitta och välj
SraRunInfo.csvfil från din dator- Klicka på Start-knappen
- stäng dialogrutan genom att trycka på Stäng knapp
- Du kan nu titta på innehållet i den här filen genom att klicka på Galaxy-Eye (Eye) – ikonen. Du kommer att se att den här filen innehåller mycket information om enskilda sra-anslutningar. I denna studie motsvarar varje anslutning en enskild patient vars prover sekvenserades.
Galaxy kan bearbeta alla 2000+ dataset men för att göra denna handledning uthärdlig måste vi välja en mindre delmängd. I synnerhet visar vår tidigare erfarenhet av dessa data två intressanta datamängder SRR11954102 och SRR12733957. Så, låt oss dra ut dem.
kommentar akta dig för skärningar
den praktiska delen nedan använder klippverktyg. Det finns två klippverktyg i Galaxy på grund av historiska skäl. I det här exemplet används verktyg med det fullständiga namnet Klipp ut kolumner från en tabell (klipp ut). Samma logik gäller dock för det andra verktyget. Det har helt enkelt ett något annorlunda gränssnitt.
hands_on Hands-on: skapa en delmängd av data
- hitta verktyg” Välj rader som matchar ett uttryck ” – verktyg i Filter och sortera avsnitt av verktygspanelen.
Tips Tips: Hitta verktyg
Galaxy kan ha en överväldigande mängd verktyg installerade. För att hitta ett specifikt verktyg skriv verktygsnamnet i verktygspanelens sökruta för att hitta verktyget.
- se till att
SraRunInfo.csvdataset som vi just laddat upp listas i fältet param-fil” Välj rader från ” i verktygsformuläret.- i fältet ”the pattern” anger du följande uttryck i uttrycket:
SRR12733957|SRR11954102. Det här är två anslutningar som vi vill hitta åtskilda av rörsymbolen||betyderor: hitta rader som innehållerSRR12733957ellerSRR11954102.- Klicka på
Execute– knappen.- detta kommer att generera en fil som innehåller två rader ( ja … en rad används också som rubrik, så det kommer att visas filen har tre rader. Det är OK.)
- Klipp ut den första kolumnen från filen med hjälp av verktyget ”Cut” – verktyg, som du hittar i Textmanipuleringsavsnittet i verktygsfönstret.
- se till att datauppsättningen som producerats av föregående steg är markerad i fältet ”fil att klippa” i verktygsformuläret.
- ändra ”avgränsad av” till
Comma- I” lista över fält”välj
Column: 1.- Hit
Executedetta kommer att producera en textfil med bara två rader:SRR12733957SRR11954102Nu när vi har identifierare av datamängder vi vill vi behöver ladda ner den faktiska sekvenseringsdata.
hämta sekvenseringsdata med snabbare nedladdning och extrahera läser i FASTQ
hands_on Hands-on: Uppgiftsbeskrivning
- snabbare nedladdning och extrahera läser i FASTQ verktyg med följande parametrar:
- ”välj Inmatningstyp”:
List of SRA accession, one per line
- parametern param-file” sra-anslutningslista ”ska peka utmatningen från verktyget” Cut ” från föregående steg.
- Klicka på knappen
Execute. Detta kommer att köra verktyget, som hämtar sekvenslästa dataset för körningarna som listades iSRAdataset. Det kan ta lite tid. Så det här kan vara en bra tid att få kaffe.- flera poster skapas i din historikpanel när du skickar in det här jobbet:
Pair-end data (fasterq-dump): Innehåller Parade dataset (om tillgängligt)Single-end data (fasterq-dump)innehåller enstaka dataset (om tillgängligt)Other data (fasterq-dump)innehåller oparade dataset (om tillgängligt)fasterq-dump loginnehåller Information om Tool executionde tre första objekten är faktiskt samlingar av datamängder. Samlingar I Galaxy är logiska grupperingar av datamängder som återspeglar de semantiska relationerna mellan dem i experimentet / analysen. I det här fallet skapar verktyget en separat samling vardera för parade slutläsningar, enstaka läsningar och andra.Se samlingarna tutorials för mer.
utforska samlingarna genom att först klicka på samlingsnamnet i historikpanelen. Detta tar dig in i samlingen och visar datamängderna i den. Du kan sedan navigera tillbaka till den yttre nivån i din historik.
När
fasterqslutar överföra data (alla rutor är gröna / färdiga) är vi redo att analysera den.nu vad?
Du kan nu analysera de hämtade data med hjälp av alla sekvensanalysverktyg och arbetsflöden i Galaxy. SRA har stöddata för alla tänkbara typer av *-seq-experiment.
om du körde den här handledningen, men hämtade dataset som du var intresserad av, se resten av GTN-biblioteket för ideer om hur du analyserar I Galaxy.
men om du hämtade datauppsättningarna som används i den här handledningens exempel ovan, är du redo att köra SARS-CoV-2-variantanalysen nedan.
Variationsanalys av SARS-Cov-2 sekvenseringsdata
i den här delen av handledningen kommer vi att utföra variantsamtal och grundläggande analys av datauppsättningarna som hämtats ovan. Vi börjar med att ladda ner Wuhan-Hu-1 SARS-CoV-2 referenssekvens, kör sedan adaptertrimning, justering och variantsamtal och slutligen titta på den geografiska fördelningen av några av de hittade varianterna.
kommentera usegalaxy.* COVID-19 analysprojekt
denna handledning använder en delmängd av data och går genomvariationsanalysavsnittet av covid19.galaxyproject.org.Uppgifterna för covid19.galaxyproject.org uppdateras kontinuerligt när nya datamängder offentliggörs.
få referensgenomdata
referensgenomdata för idag är för SARS-CoV-2,” allvarligt akut respiratoriskt syndrom coronavirus 2 isolera Wuhan-Hu-1, komplett genom”, med anslutnings-ID för NC_045512.2.
dessa data är tillgängliga från Zenodo med följande länk.
hands_on Hands-on: hämta referensgenomdata
importera följande fil till din historik:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gztips: importera via länkar
- kopiera länkplatsen
- öppna Galaxy Upload Manager (galaxy-upload längst upp till höger på verktygspanelen)
- välj Klistra in/hämta data
- klistra in länken i textfältet
- tryck på Start
- stäng fönstretsom standard använder Galaxy webbadressen som namn, så byt namn på filerna med ett mer användbart namn.
Adapter trimning med fastp
Ta bort sekvenseringsadaptrar förbättrar inriktningar och variant ringer. fastp tool kan automatiskt upptäcka allmänt använda sekvenseringsadaptrar.
hands_on Hands-on: Uppgiftsbeskrivning
- fastp-verktyg med följande parametrar:
- ”enstaka eller parade läser”:
Paired Collection
- param-fil ”Välj Parade samlingar”:
list_paired(utmatning av snabbare nedladdning och extrakt läser i FASTQ-verktyget)- I ”Output Options”:
- ”Output JSON report”:
Yesjustering med karta med BWA-mem
bwa-mem Tool är en allmänt använd sekvensinställare för Kortlästa sekvenseringsdataset som de vi analyserar i denna handledning.
hands_on Hands-on: Justera sekvensering läser till referensgenom
- karta med BWA-MEM-verktyg med följande parametrar:
- ” kommer du att välja ett referensgenom från din historik eller använda ett inbyggt index?”:
Use a genome from history and build index
- param-fil ”använd följande dataset som referenssekvens”:
output(Indataset)- ”enkel eller parad-end läser”:
Paired Collection
- param-fil ”välj en parad samling”:
output_paired_coll(utmatning av fastp-verktyg)- ”Ställ in läsgrupper information?”:
Do not set- ”Välj analysläge”:
1.Simple Illumina modeta bort dubbletter med Markduplikat
markduplikatverktyget tar bort dubbla sekvenser som härrör från bibliotekets förberedelse artefakter och sekvensering artefakter. Det är viktigt att ta bort dessa artefakt sekvenser för att undvika artificiell överrepresentation av enstaka molekyl.
hands_on Hands-on: ta bort PCR dubbletter
- MarkDuplicates verktyg med följande parametrar:
- param-file ”välj SAM/Bam dataset eller dataset collection”:
bam_output(utmatning av Karta med BWA-mem-verktyg)- ”om sant skriv inte dubbletter till utdatafilen istället för att skriva dem med lämpliga flaggor”:
Yesgenerera justeringsstatistik med samtools stats
efter duplikatmarkeringssteget ovan kan vi generera statistik om justeringen vi har genererat.
hands_on Hands-on: Generera justeringsstatistik
- samtools stats tool med följande parametrar:
- param-file ”BAM file”:
outFile(utmatning av MarkDuplicates tool)- ”Set coverage distribution”:
No- ”Output”:
One single summary file- ”filtrera efter Sam-flaggor”:
Do not filter- ”använd en referenssekvens”:
No- ”filtrera efter regioner”:
NoRealign läser med lofreq viterbi
Realign läser verktyget korrigerar feljusteringar kring Infogningar och borttagningar. Detta krävs för att exakt upptäcka varianter.
hands_on Hands-on: Realign läser runt indels
- Realign läser med lofreq verktyg med följande parametrar:
- param-fil ”läser till realign”:
outFile(utmatning av MarkDuplicates verktyg)- ”Välj källan för referensgenomet”:
History
- param-fil ”referens”:
output(Input dataset)- i” Avancerade alternativ”:
- ” hur hanterar du baskvaliteter på 2?”:
Keep unchangedLägg till indel-kvaliteter med lofreq infoga indel-egenskaper
detta steg lägger till indel-kvaliteter i vår justeringsfil. Detta är nödvändigt för att ringa varianter med hjälp av Samtalsvarianter med lofreq tool
hands_on Hands-on: Lägg indel kvaliteter
- infoga indel kvaliteter med lofreq verktyg med följande parametrar:
- param-fil ”läser”:
realigned(utgång av Realign läser verktyg)- ”Indel beräkning approach”:
Dindel
- ”Välj källan för referensgenomet”:
History
- param-fil ”referens”:
output(indataset)anropsvarianter med lofreq Call variants
är vi nu redo att ringa varianter.
hands_on Hands-on: Samtalsvarianter
- Samtalsvarianter med lofreq-verktyg med följande parametrar:
- param-file ”Input läser i BAM-format”:
output(utmatning av Insert Indel qualities tool)- ”Välj källa för referens genom”:
History
- param-fil ”referens”:
output(indataset)- ”Ring varianter över”:
Whole reference- ”typer av varianter att ringa”:
SNVs and indels- ”Variant anropande parametrar”:
Configure settings
- I ”täckning”:
- ”Minimal täckning”:
50- I ”Base-calling”:
- ”minsta baseq”:
30- ”minsta baseq för alternativa baser”:
30- i ”mapping qualityy
20- ”variantfilterparametrar”:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)utgången från detta steg är en samling VCF-filer som kan visualiseras i en genombläddrare.
kommentera varianteffekter med SnpEff eff:
Vi kommer nu att kommentera varianterna vi ringde i föregående steg med den effekt de har på SARS-CoV-2-genomet.
hands_on Hands-on: kommentera varianteffekter
- SnpEff eff: verktyg med följande parametrar:
- param-fil ”Sekvensändringar (SNPs, MNPs, InDels)”:
variants(utmatning av Samtalsvarianter verktyg)- ”Output format”:
VCF (only if input is VCF)- ”skapa CSV-rapport, användbar för nedströmsanalys (- csvStats)”:
Yes- ”Annotation options”: `
- ”filter output”: `
- ”filtrera bort specifika effekter”:
Noutsignalen från detta steg är en VCF-fil med tillagda varianteffekter.
Skapa tabell med varianter med SnpSift-Extraktfält
vi väljer nu olika effekter från VCF och skapar en tabellfil som är lättare att förstå för människor.
hands_on Hands-on: Skapa tabell med varianter
- SnpSift Extract Fields tool med följande parametrar:
- param-file ”Variant inmatningsfil i VCF-format”:
snpeff_output(utmatning av SnpEff eff: tool)- ”fält att extrahera”:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF.IMPACT EFF.FUNCLASS EFF.EFFECT EFF.GENE EFF.CODON- ”multiple field separator”:
,- ”tomt fält text”:
.Vi kan inspektera utdatafilerna och se om varianter i den här filen också beskrivs i en observerbar anteckningsbok som visar den geografiska fördelning av SARS-CoV-2 variantsekvenser
intressanta varianter inkluderar C till t-varianten vid position 14408 (14408c/t) i srr11772204, 28144t/C i srr11597145 och 25563g/t i srr11667145.
sammanfatta data med MultiQC
Vi kommer nu att sammanfatta vår analys med MultiQC, vilket genererar en vacker rapport för våra data.
hands_on Hands-on: sammanfatta data
- MultiQC-verktyg med följande parametrar:
- I ”resultat”:
- param-repeat ”infoga resultat”
- ”vilket verktyg användes generera loggar?”:
fastp
- param-fil ”utmatning av fastp”:
report_json(utmatning av fastp-verktyg)- param-upprepa ”infoga resultat”
- ”vilket verktyg användes generera loggar?”:
Samtools
- I ”Samtools output”:
- param-upprepa” infoga Samtools output ”
- ” typ av Samtools output?”:
stats
- param-fil ”Samtools statistik utgång”:
output(utmatning av Samtools statistikverktyg)param-upprepa ”infoga resultat”
- ”vilket verktyg användes generera loggar?”:
Picard
- I ”Picard-utgång”:
- param-upprepa” infoga Picard-utgång ”
- ” typ av Picard-utgång?”:
Markdups- param-fil ”Picard-utgång”:
metrics_file(utmatning av MarkDuplicates tool)- param-upprepa ”infoga resultat”
- ”vilket verktyg användes generera loggar?”:
SnpEff
- param-fil” utmatning av SnpEff”:
csvFile(utmatning av SnpEff eff: verktyg)slutsats
grattis, du vet nu hur man importerar sekvensdata från sra och hur man kör en exempelanalys på dessa dataset.
keypoints nyckelpunkter
sekvensdata i Sra kan importeras direkt till Galaxy
Vanliga frågor
har du frågor om denna handledning? Kolla in FAQ-sidan för Variantanalysämnet för att se om din fråga är listad där. Om inte, Ställ din fråga på GTN Gitter-kanalen eller Galaxy Help Forum
användbar litteratur
ytterligare information, inklusive länkar till dokumentation och originalpublikationer, om verktyg, analystekniker och tolkning av resultat som beskrivs i denna handledning finns här.
Feedback
använde du detta material som instruktör? Känn dig fri att ge oss feedback om hur det gick.
citerar denna handledning
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, 2021 från NCBIS Sekvensläsningsarkiv (Sra) till Galaxy: SARS-CoV-2-variantanalys (Galaxy-träningsmaterial). / utbildning-material/ämnen/variant-analys/tutorials/sars-cov-2 / handledning.html Online; nås idag
- Batut et al., 2018 Community-Driven dataanalys utbildning för biologi cellsystem 10.1016 / j. cels.2018.05.012
details BibTeX
@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "2021", month = "03", day = "23" url = "\url{/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = ""}@article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems}}