aminosyror, nukleotider eller någon annan evolutionär karaktär ersätts avandra i viss takt. Föreställ dig till exempel en evolutionär sekvens med tremöjliga tillstånd, A, B och C. Om substitutionsmodellen är tidsreversibel kommer det att finnas tre övergångshastigheter, a<>B, B<>C och a<>C.
Antag att räntorna är 1, 1 respektive 0 i substitutionsenheter per 100tecken per tidsenhet. Efter en tidsenhet, i en 300 tecken longsequence ursprungligen sammansatt lika av As, Bs och Cs, förväntar vi oss att habeen en A till B substitution och en B till C substitution. Om vi jämför två homologa sekvenser i levande organismer, eftersom en tidsenhet har passerat för båda sekvenserna, skulle vi förvänta oss två A till B och två B till Csubstitutioner mellan dagens sekvenser.
oavsett hur länge vi kör denna process för kommer det aldrig att bli en direktplacering av A med C. Det kommer aldrig att bli en A till C-substitution under aso-kallad infinite sites-modell, där inte mer än en substitution kan förekomma på en enda plats.men eftersom A till B och B till C substitutioner är vanliga, under en ändlig sitesmodell så småningom B kommer att ersättas med C på en plats där A tidigare ersattes av B. Denna indirekta ersättning av A med C (eller motsvarande i atime-reversibel Modell, C av A) blir mer sannolikt ju längre tidsperiodenseparerar de homologa sekvenserna.
jag simulerade sekvensutveckling baserat på ovanstående scenario, körsimulation för 10 tidsenheter. Från denna substitution observerade jag följande räkningar för varje webbplatsmönster:
| A | B | C | |
|---|---|---|---|
| A | 91 | 9 | 0 |
| B | 5 | 86 | 9 |
| C | 0 | 9 | 91 |
inom denna relativt korta varaktighet verkar det inte som om någon a<>csubstitutions har inträffat. Men när jag reran simuleringen för 100 unitsof tid:
| A | B | C | |
|---|---|---|---|
| A | 55 | 35 | 10 |
| B | 29 | 36 | 35 |
| C | 20 | 36 | 44 |
som du kan se har många ”a” – tecken ersatts med ”C” och vice versa. Mer allmänt, under en ändlig platser modell flera substitutionerorsaka fördelningen av plats mönster räknas att bli mycket plattare beyondsimply öka andelen off-diagonal i förhållande till diagonala räkningar.Pam-och BLOSUM-poängmatriserna står för flera substitutioner iradikalt olika sätt.
PAM-matriserna för aminosyror, tillsammans med förkortningarna med en bokstav som används för genetiskt kodade aminosyror, utvecklades av MargaretDayhoff. De publicerades ursprungligen 1978 och baserade på proteinsekvenserna Dayhoff hade sammanställt sedan 1960-talet, publicerad som theAtlas of Protein Sequence and Structure.
namnet PAM kommer från ”punkt accepterad mutation” och hänvisar tillplacering av en enda aminosyra i ett protein med en annan aminosyra.Dessa mutationer identifierades genom att jämföra mycket liknande sekvenser med minst 85% identitet, och det antas att alla observerade substitutioner var resultatet av en enda mutation mellan förfädersekvensen och en av dagens sekvenser.
Pam definierar också en tidsenhet, där 1 PAM är den tid då 1/100 aminosyror förväntas genomgå en mutation. PAM1-sannolikhetsmatrisen visarsannolikheten för att aminosyran vid kolumn j ersätts av aminosyran vid rad i. den beräknades från Dayhoffs Pam-räkningar, och omskalad tobe 1 Pam-tidsenhet. Som du kan se är de Off-diagonala sannolikheterna ipam1-matrisen alla mycket små (alla element skalades med 10 000 förläsbarhet):
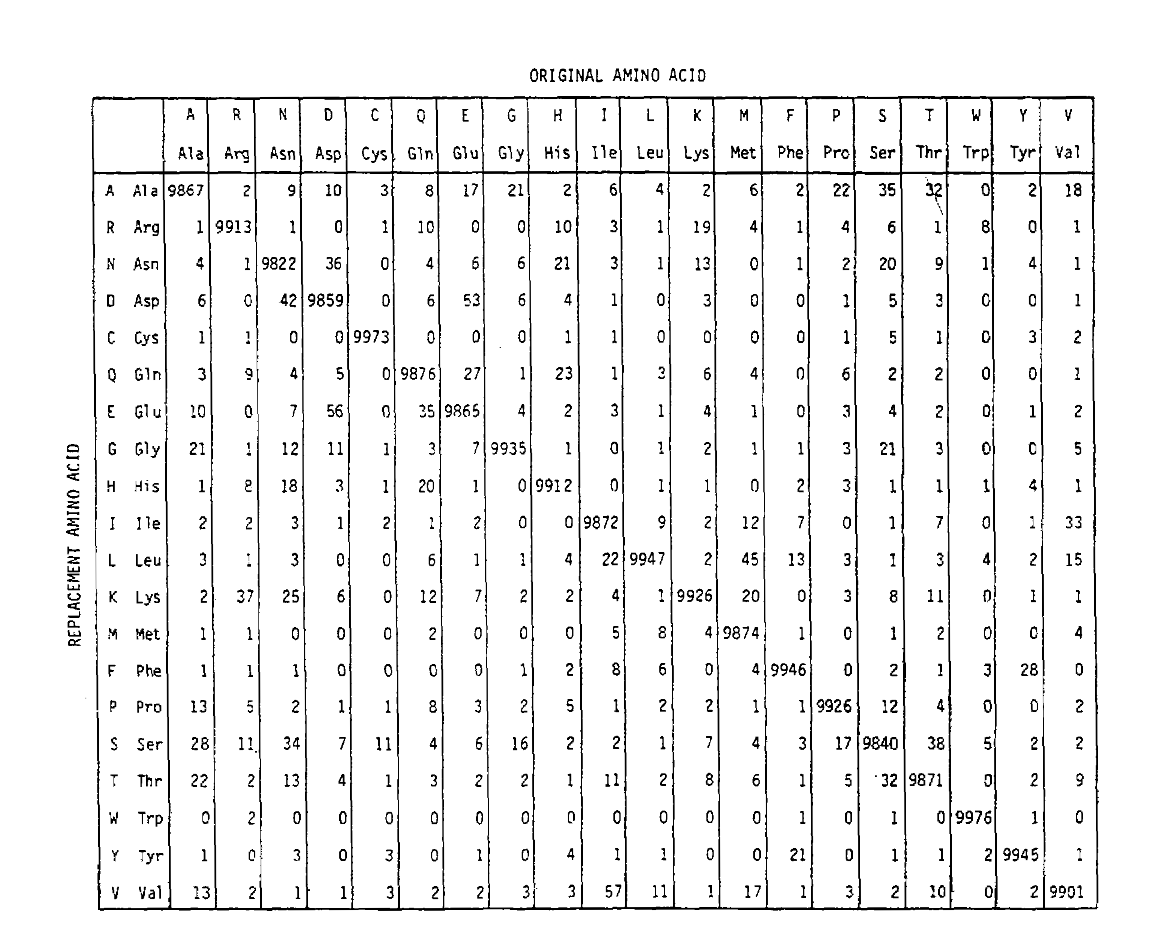
för att beräkna aminosyrautbytessannolikheterna för längre tidurationer kan matrisen multipliceras med sig själv motsvarandeantal gånger. Således PAM250 sannolikhetsmatris, som beskriver thereplacement sannolikheter ges 250 PAM tidsenheter, härleddes genom höjningpam1 sannolikhetsmatris till kraften 250 (alla element skalas av 100for läsbarhet):
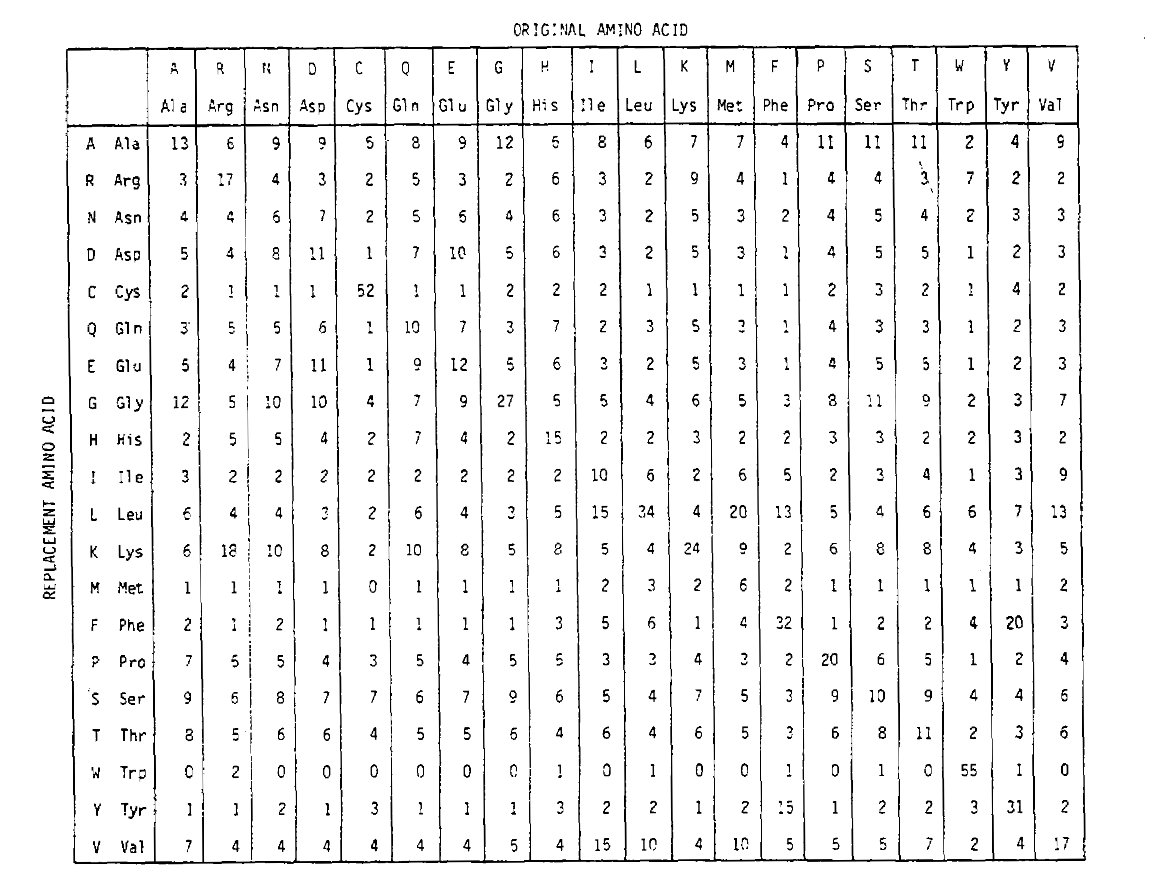
ersättningssannolikheterna härledda med hjälp av denna exponentiering accountly för flera substitutioner. Inte bara är off-diagonalsannolikheterna proportionellt större som du förväntar dig under en längre tid, men de är plattare. Till exempel är sannolikheten för en valin (V)till isoleucin (I) – ersättning 33 kg större än en v till histadin (H) – ersättning i PAM1-matrisen, men endast 4,5 kg större i PAM250-matrisen.
Poängmatriser kan sedan beräknas från sannolikhetsmatriserna ochobserverade basfrekvenser.
BLOSUM matrices, utvecklad av Steven och Jorja Henikoff och publicerad i 1992, tar ett helt annat tillvägagångssätt. Medan PAM implicit tillämpar en stationär ändlig platsmodell av evolution med matrisexponentiering, behandlas effekten av flera substitutioner implicit i BLOSUM genom att konstruera olika poängmatriser för olika tidsskalor.
inom multipla sekvensinriktningar av homologa sekvenser identifieras konserverade sammanhängande block av aminosyror. Inom varje block grupperas multiplesequences när deras parvisa genomsnittliga sekvensidentitet är högre än någon tröskel. Tröskeln är 80% för blosum80-matrisen, 62% för BLOSUM62, 50% för BLOSUM50 och så vidare.
detta betyder att för BLOSUM80 kommer block att ha genomsnittliga parvisa identiteter inte större än 80%, för BLOSUM62 inte större än 62%, et cetera.
Aminosyraersättningssannolikheter för homologa sekvenser beräknas från parvisa jämförelser mellan kluster. Dessa sannolikheter kommer att vara resultatet av enkla och multipla substitutioner, med flera substitutioner som har större inflytande på större evolutionära avstånd. Därför kommer scorematrices genererade från parvisa jämförelser mellan kluster av i genomsnittstörre avstånd, som BLOSUM50-matrisen, naturligtvis att redogöra för denstörre effekten av flera substitutioner.
Även om de tar olika vägar, är de slutliga BLOSUM-och PAM-poängmatriserna faktiskt ganska lika. Enligt Henikoff och Henikoff, followingPAM och BLOSUM matriser är jämförbara:
| PAM | BLOSUM |
|---|---|
| PAM250 | BLOSUM45 |
| PAM160 | BLOSUM62 |
| PAM120 | BLOSUM80 |
For more information on PAM (Dayhoff) and BLOSUM matrices, see kapitel 2 avbiologisk sekvensanalys av Durbin et al. och Wikipedia.
Uppdatering 13 oktober 2019: för ett annat perspektiv på substitutionsmatriser, se avsnittet ”omvägar” i slutet av kapitel 5 i Bioinformatikalgoritmer (2: A eller 3: e upplagan) av Compeau och Pevzner.